Diffuse scattering contains two-body information of the system under investigation and is thus a valuable source how atoms or molecules interact. In contrast Bragg scattering can only reveal average one-body information such as atomic positions, site occupancies and thermal ellipsoids. In this chapter the concept of correlations as a measure for those two-body interactions will be introduced. Although diffuse scattering contains only information about two-body interactions the concepts described here can easily be extended to include multi-site correlations. It should be noted that although these multi-site interactions do not show up in the diffraction pattern directly, they can have a constraining influence on two-body interactions and thus effecting the diffraction pattern. However, DISCUS is currently limited to the calculation of two-body interaction averages.
We will talk about atom types in the following section, however, all correlation related commands are available for molecules as well. To work with molecules use the command 'mode mole' and specify molecule types rather than atom types or names as parameters for the commands. The command 'homo' (see section 8.1) can also be used to determine the homgeniety of correlations within the crystal.
First occupational correlations will be discussed. One definition of the correlation coefficient cij between a pair of sites i and jbased on a statistical definition of correlation [28] is given in equation 8.1.
Pij is the joint probability that both sites i and j are occupied
by the same atom type and ![]() is its overall occupancy. Negative
values of cij correspond to situations where the two sites i and jtend to be occupied by different atom types while positive values
indicate that sites i and j tend to be occupied by the same atom
type. A correlation value of zero describes a random distribution. The
maximum negative value of cij for a given concentration
is its overall occupancy. Negative
values of cij correspond to situations where the two sites i and jtend to be occupied by different atom types while positive values
indicate that sites i and j tend to be occupied by the same atom
type. A correlation value of zero describes a random distribution. The
maximum negative value of cij for a given concentration ![]() is
is
![]() (Pij=0), the maximum positive value is +1
(
(Pij=0), the maximum positive value is +1
(
![]() ). This definition of correlations can easily be
converted to the Warren-Cowley short-range order parameter
). This definition of correlations can easily be
converted to the Warren-Cowley short-range order parameter
![]() [5].
As an example the correlations
[5].
As an example the correlations
![]() and
and
![]() of the disordered structure shown in figure 8.1 are
calculated using the DISCUS macro file listed below:
of the disordered structure shown in figure 8.1 are
calculated using the DISCUS macro file listed below:
1 read
2 stru chem.1.stru
3 #
4 chem
5 #
6 set mode,quick,periodic
7 #
8 set vec,1,1,1, 1, 0, 0
9 set vec,2,1,1,-1, 0, 0
10 set vec,3,1,1, 0, 1, 0
11 set vec,4,1,1, 0,-1, 0
12 #
13 set vec,5,1,1, 1, 1, 0
14 set vec,6,1,1,-1, 1, 0
15 set vec,7,1,1, 1,-1, 0
16 set vec,8,1,1,-1,-1, 0
17 #
18 set neig,vec,1,2,3,4
19 set neig,add
20 set neig,vec,5,6,7,8
21 #
22 corr occ,zr,void
23 #
24 exit
The macro starts with the reading of the disordered structure (lines 1-2).
After the 'chem' sublevel is entered (line 4) periodic crystal boundaries
are selected (line 6). The parameter 'quick' selects a faster neigbouring
finding algorithm which only works for crystals arranged in the DISCUS storage order (see section 2.2). Note that
![]() stands for the nearest neighbour correlations in all four
symmetrically equivalent <10> directions of the two dimensional cubic test
crystal, i.e. c10,
stands for the nearest neighbour correlations in all four
symmetrically equivalent <10> directions of the two dimensional cubic test
crystal, i.e. c10,
![]() ,
c01 and
,
c01 and
![]() .
All eight neigbouring directions for
.
All eight neigbouring directions for
![]() and
and
![]() are defined as vectors 1 to 8 in
lines 8 to 16 of the macro file. Next vectors 1 to 4 are grouped as
neighbouring definition for
are defined as vectors 1 to 8 in
lines 8 to 16 of the macro file. Next vectors 1 to 4 are grouped as
neighbouring definition for
![]() (line 18) and vectors
5 to 8 for
(line 18) and vectors
5 to 8 for
![]() (line 20). The command 'set neig,add'
(line 19) stores the current neighbouring definition and allows the
definition of a new one. Finally the correlations for the defined
neighbouring directions are calculated (line 22). The screen output for
the disordered structure shown in figure 8.1 looks like this:
(line 20). The command 'set neig,add'
(line 19) stores the current neighbouring definition and allows the
definition of a new one. Finally the correlations for the defined
neighbouring directions are calculated (line 22). The screen output for
the disordered structure shown in figure 8.1 looks like this:
Calculating correlations
Atom types : A = ZR and B = VOID
Neig. AA AB BB # pairs correlation
-----------------------------------------------------------------
1 50.49 % 48.99 % .51 % 40000 -.3061
2 71.04 % 7.89 % 21.07 % 40000 .7897
The program lists the probabilities for AA, AB and BB pairs and the
corresponding correlations cij. Here the value for
![]() is negative, i.e. vacancy neighbours in <10> directions tend
to be avoided. Neighbouring vacancies in <11> direction on the other
hand are much more likely compared to a random vacancy distribution
indicated by the large positive value of
is negative, i.e. vacancy neighbours in <10> directions tend
to be avoided. Neighbouring vacancies in <11> direction on the other
hand are much more likely compared to a random vacancy distribution
indicated by the large positive value of
![]() .
This
leads to the large areas with a doubled unit cell and a Zr concentration of
0.5 as seen in section 8.1.
.
This
leads to the large areas with a doubled unit cell and a Zr concentration of
0.5 as seen in section 8.1.
The correlation coefficient cij for displacement correlations between two sites i and j is defined as:
Here xi is the displacement of the atom on site i from the average
position in a given direction and
![]() stands for the
average over the crystal. Again a negative value describes a situation
where the pairing of corresponding displacements are less likely than in
a crystal with random displacements whereas a positive value indicates a
larger than random probability. The definition of neighbours is
identical to the example in the previous section. Additionally the
command 'set neig,dir' is used to determine the displacement direction
to be used. Note that the displacement direction for the two sites
i and j is not necessarily the same, e.g. one could be interested in
the correlation between the x-displacement on one site and the y-displacement
on the neighbouring site.
stands for the
average over the crystal. Again a negative value describes a situation
where the pairing of corresponding displacements are less likely than in
a crystal with random displacements whereas a positive value indicates a
larger than random probability. The definition of neighbours is
identical to the example in the previous section. Additionally the
command 'set neig,dir' is used to determine the displacement direction
to be used. Note that the displacement direction for the two sites
i and j is not necessarily the same, e.g. one could be interested in
the correlation between the x-displacement on one site and the y-displacement
on the neighbouring site.
In the previous sections a correlation cij for a given pair of neighbouring atoms was computed. An interesting information, however, is how these correlations extend within the crystal. The program DISCUS allows the calculation of correlation fields for occupational and displacement correlations (command: 'field').
Again taking the disordered structure displayed in figure 8.1
as an example, the correlation fields in <10> and <11> direction
are shown in figure 8.3. This figure shows the correlation
between a site i and sites separated by integer multiples of the
vectors used in the neighbor definitions. The neighbouring definitions
are the same as in the example for a single correlation coefficient in section
8.3.1. The first neighbour correlation is negative for
![]() and positive for
and positive for
![]() as in
section 8.3.1. The correlation
as in
section 8.3.1. The correlation
![]() is
decaying as a function of the distance within the crystal, giving a measure
of the extension of the area with preferred <11> vacancy neighbours.
Eventually the value gets negative for distances above about 26 times the
is
decaying as a function of the distance within the crystal, giving a measure
of the extension of the area with preferred <11> vacancy neighbours.
Eventually the value gets negative for distances above about 26 times the
![]() vector which can be understood taking the size
of the two phase regions (figure 8.1) into account.
The absolute value of correlation
vector which can be understood taking the size
of the two phase regions (figure 8.1) into account.
The absolute value of correlation
![]() decays as well
but oscillates between negative an positive values. This can be understood
by thinking of a perfect ABAB sequence. All odd neighbours (i.e. 1,3,5,
...) are AB or BA resulting in a negative correlation whereas even
neighbours (i.e. 2,4,6, ...) are AA or BB giving a positive correlation.
decays as well
but oscillates between negative an positive values. This can be understood
by thinking of a perfect ABAB sequence. All odd neighbours (i.e. 1,3,5,
...) are AB or BA resulting in a negative correlation whereas even
neighbours (i.e. 2,4,6, ...) are AA or BB giving a positive correlation.
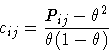
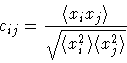
![\includegraphics[scale=0.50, angle=0]{chem.3.eps}](img134.gif)