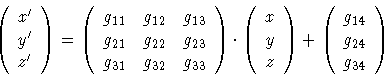A unit cell or a whole structure is read from file by the command 'read'. The format of these two file types is identical. If a unit cell is read, the contents of the file is regarded as the asymmetric unit of the unit cell. The space group information is used to generate the whole unit cell. If, on the other hand, a structure is read, the contents of the file is taken as it is.
A keyword controlled structure file format is used allowing more flexibility, e.g. the use of molecules. However, DISCUS is capable to read both the old and the new structure files automatically recognizing the correct format. Furthermore the command 'format nokey' in the 'save' segment allows the user to save a structure using the old format (see section 2.3).
The new DISCUS structure file is a simple text file starting with a section with keywords setting parameters like lattice constants or space group followed by a section with the actual atoms. The sequence of keywords is arbitrary with two exceptions, the first line must contain the keyword 'title' setting a title and the last keyword must be atoms followed by a list of atoms and/or molecule definitions (see section 2.1.2). Each atom within the asymmetric unit or complete structure is defined by its name (e.g. ZR), the fractional coordinates (x,y,z) and an isotropic temperature factor B. A list of valid keywords within a structure file is given in table 2.1.
|
Keywords are processed the same way as normal commands and parameters have to be separated by commas ','. An example input file for cubic zirconia (ZrO2) is shown below.
title Structure of cubic ZrO2
spcgr Fm-3m
cell 5.14,5.14,5.14,90.,90.,90.
atoms
ZR 0.00 0.00 0.00 0.5
O 0.25 0.25 0.25 1.0
The first line is the required 'title' line describing the structure. The
next line specifies the space group
![]() .
The symbols used
should be the Hermann-Mauguin symbols used in International Tables for
Crystallography Vol. A [33]. A center of inversion as in this
example should be given as '-' sign immediately preceding the axis.
Lattice types need to be given as capital characters, mirror planes as
small characters. Monoclinic cell choices 2,3 or unique c-axis will be
assumed if the corresponding non standard Hermann-Mauguin symbol is used.
DISCUS checks the given space group symbol for contradictions with
the lattice constants and in case of an error the unit cell is not read. A
complete list of valid space group symbols is part of the online help and
can be accessed via 'help space' from the DISCUS command line. The
next line in the example above gives the lattice constants of
a=b=c=5.14Å and
.
The symbols used
should be the Hermann-Mauguin symbols used in International Tables for
Crystallography Vol. A [33]. A center of inversion as in this
example should be given as '-' sign immediately preceding the axis.
Lattice types need to be given as capital characters, mirror planes as
small characters. Monoclinic cell choices 2,3 or unique c-axis will be
assumed if the corresponding non standard Hermann-Mauguin symbol is used.
DISCUS checks the given space group symbol for contradictions with
the lattice constants and in case of an error the unit cell is not read. A
complete list of valid space group symbols is part of the online help and
can be accessed via 'help space' from the DISCUS command line. The
next line in the example above gives the lattice constants of
a=b=c=5.14Å and
![]() degrees. Note that DISCUS requires all six values to be given. The keyword 'atoms' in the
example file which must be the last keyword starts the section with the
list of atoms. Here zirconium occupies site 4(a) on (0,0,0) and oxygen is
on 8(f) at (
degrees. Note that DISCUS requires all six values to be given. The keyword 'atoms' in the
example file which must be the last keyword starts the section with the
list of atoms. Here zirconium occupies site 4(a) on (0,0,0) and oxygen is
on 8(f) at (
![]() ). The isotropic
temperature factors for Zr and O are set to 0.5 Å2 and 1.0 Å2respectively.
). The isotropic
temperature factors for Zr and O are set to 0.5 Å2 and 1.0 Å2respectively.
Additional generators can be defined through the optional 'generator' keyword. These generators act identical to the generators defined through the space group symbol. All previously generated copies of the atoms in the asymmetric unit are copied by this generator, and will in turn be copied by any generators following later. Since these additional generators are applied after the space group generators, you can use these generators to create non-standard groups or to create a set of symmetries that does not from a group. The syntax of the 'generator' keyword is as follows:
generator g11,g12,g13,g14, g21,g22,g23,g24, g31,g32,g33,g34
Copies of an atom at (x,y,z) will be calculated using the following equation:
Additional symmetry operations can be defined through the optional 'symmetry' keyword. These symmetry operations act different than the generators described above which are defined through the space group symbol or listed as additional generators. The symmetry operations copy only those atoms created by the generators. In contrast to generators they do not act on copies of the atoms created by previous symmetry operations. Both keywords 'generator' and 'symmetry' define the symmetry operation in a similar way using 12 parameters as shown in equation 2.1
The following example shall illustrate the difference between generators and additional symmetry operations. The following two generators
generator 1,0,0,0.5, 0,1,0,0.5, 0,0,1,0.0
generator 1,0,0,0.5, 0,1,0,0.0, 0,0,1,0.5
would create the following copies of an atom at (0,0,0):
![]() and
and
![]() .
In contrast similar symmetry operations
.
In contrast similar symmetry operations
symmetry 1,0,0,0.5, 0,1,0,0.5, 0,0,1,0.0
symmetry 1,0,0,0.5, 0,1,0,0.0, 0,0,1,0.5
will only generate the following two copies of an atom at (0,0,0):
![]() and
and
![]() since the symmetry operations will not act on previously generated
copies of the atom at (0,0,0). The second symmetry operation copies
only the atom at (0,0,0), not the atom at
since the symmetry operations will not act on previously generated
copies of the atom at (0,0,0). The second symmetry operation copies
only the atom at (0,0,0), not the atom at
![]() ,
since this atom was created by the
previous symmetry operation.
,
since this atom was created by the
previous symmetry operation.
The new ![]() ]
]![]() keyword controlled structure file format of DISCUS
allows the definition of molecules using the 'molecule' keyword. This
keyword is allowed anywhere between the atoms of the unit cell file. It
marks the beginning of a group of atoms that are grouped to form a
molecule. The individual atoms are listed in the usual way (see section
2.1.1). The keyword 'molecule end' signals the end of a
molecule. All atoms still listed in the unit cell file are treated as
individual atoms. The molecule related keywords are listed in table
2.2.
keyword controlled structure file format of DISCUS
allows the definition of molecules using the 'molecule' keyword. This
keyword is allowed anywhere between the atoms of the unit cell file. It
marks the beginning of a group of atoms that are grouped to form a
molecule. The individual atoms are listed in the usual way (see section
2.1.1). The keyword 'molecule end' signals the end of a
molecule. All atoms still listed in the unit cell file are treated as
individual atoms. The molecule related keywords are listed in table
2.2.
|
The internal symmetry of the molecule can be specified using the 'generator' and 'symmetry' sub-keywords. The generators are internal symmetry operations of the molecule. DISCUS compares the lists of atoms created by the space group and by the molecule generators. Identical sections are linked to one molecule. Atoms created by other symmetry operations, e.g. lattice centering will form a new molecule of the same type. The generators of the molecule symmetry should be the generators that create the site symmetry. See the section on site symmetry in the International Tables [33] for further details. As in the previous section, symmetry operations will only act on the 'original' atoms of the molecule whereas generators will operate on previously generated copies of atoms as well. The following example structure file contains a water (H2O) molecule in a structure with the space group Cmm2.
title Water in Cmm2
spcgr Cmm2
cell 10.0,10.0,10.0, 90.0,90.0,90.0
atoms
molecule
molecule gene, -1,0,0,0, 0,1,0,0, 0,0,1,0
O 0.00 0.20 0.00 0.1
H 0.13 0.17 0.00 0.2
molecule end
The first four lines of this example file are similar to the previous example and define title, space group and lattice constants. In the 'atoms' section, however, one oxygen and one hydrogen atom define the water molecule between the 'molecule' and 'molecule end' keywords. The second hydrogen atom for the H2O molecule is generated by a yz-mirror plane defined by the 'gene' sub-keyword. The mirror plane goes through the origin of the molecule which is defined as the first atom in the molecule list, here oxygen. The coordinates of the four created H2O molecules per unit cell in the given space group Cmm2 are shown below.
Molecule Number: 1 Type: 1
Name Number x y z B
O(1) 1 .000000 .200000 .000000 .100000
H(2) 5 .130000 .170000 .000000 .200000
H(2) 11 -.130000 .170000 .000000 .200000
Molecule Number: 2 Type: 1
Name Number x y z B
O(1) 2 .500000 .700000 .000000 .100000
H(2) 6 .630000 .670000 .000000 .200000
H(2) 12 .370000 .670000 .000000 .200000
Molecule Number: 3 Type: 1
Name Number x y z B
O(1) 3 .000000 .800000 .000000 .100000
H(2) 7 -.130000 .830000 .000000 .200000
H(2) 9 .130000 .830000 .000000 .200000
Molecule Number: 4 Type: 1
Name Number x y z B
O(1) 4 .500000 .300000 .000000 .100000
H(2) 8 .370000 .330000 .000000 .200000
H(2) 10 .630000 .330000 .000000 .200000
There is one important restriction how molecules are defined in DISCUS: The first atom of any molecule defines the origin of the molecule used by various subsequent commands. In case the origin lies on a symmetry element of the space group it must be located at the point of highest symmetry of the molecule. If the structure does not have an atom at this site you must include a 'void' on this site. This could be the case e.g. if you have an empty triangle on a threefold axis.
Alternatively to defining a molecule as in the example above, the command 'molecule content' and 'molecule atoms' might be used to define molecule types and the corresponding list of atom indices belonging to that molecule. This procedure is used to be able to save structures containing molecules since the order of the atoms required by various DISCUS functions might prevent storing atoms in groups belonging to a particular molecule. Check section 2.3 for more details about saving structures.