STRUCTURAL BIOLOGY ON THE SODIUM PUMP: A COMBINED
APPROACH LEADING TO A FULL CHARACTERIZATION OF THE CATALYTIC DOMAIN.
K. Hofbauerová1,2, V. Kopecký Jr.1,2,3, R. Ettrich4,
M. Kubala2,3, J.
Teisinger2,
O. Ettrichová2,4
and E. Amler2
1Department of Biochemistry, Faculty
of Sciences, Charles University, Albertov 2030, CZ-128 40 Prague 2, Czech Republic, email:
hofbauer@biomed.cas.cz
2Institute of Physiology, Czech
Academy of Sciences, Vídeňská 1083, CZ-142 20 Prague, Czech Republic
3Institute of Physics, Charles
University, Ke Karlovu 5, CZ-121 16 Prague 2, Czech Republic
4Laboratory of High
Performance Computing, Institute of Physical Biology USB and Institute of
Landscape Ecology AS CR, University of South Bohemia, Zámek 136, CZ-373
33 Nové Hrady, Czech Republic
Na+/K+-ATPase (EC 3.6.1.37) is an integral membrane protein.
This enzyme consists of two subunits, the catalytic a subunit
and the associated glycoprotein b subunit [1]. The a subunit contains 10 transmembrane segments with a large cytoplasmic
loop, which is located between helices H4 and H5, where
the ATP binding site and the phosphorylation site are localized [2]. This H4–H5 loop was
shown to preserve a rigid and self-supporting structure [3] and is
able to bind nucleotide triphosphates [4]. On the
basis of the recently solved tertiary structure of Ca2+-ATPase,
the complete 3D structure of the large cytoplasmic loop between the fourth and
fifth transmembrane segment (H4–H5 loop) of
the a subunit of Na+/K+-ATPase, beginning with Lys354 and ending
with Lys773, was modeled using the method of
restraint-based comparative modeling [5]. Due to the relatively high degree of
homology of more than 50% with respect to the cytoplasmic loop of Ca2+-ATPase and the sufficient amount of background
information, contributed by different labelling techniques of certain residues
and kinetic studies, it was possible to create a model in which all amino acids
were modeled convincingly. We have shown that the ATP binding site and the
phosphorylation site are located on two different, well-separated domains,
which together form the large cytoplasmic loop. For these domains we propose to
use the following names: domain P comprises the N-terminal and C-terminal ending
of the H4–H5 loop and
contains Asp369, the residue of phosphorylation surrounded by
a highly negatively charged surface; and domain N, which binds nucleotides. We
also show that there is evidence for only one ATP binding site on the N domain
of the H4–H5 loop, and
we were able to specify Cys549, Phe548, Glu505, Lys501, Gln482, Lys480, Ser477, Phe475 and Glu446 as parts
of the ATP binding site with Lys501 located in
the depth of the positively charged binding pocket (Fig. 1).
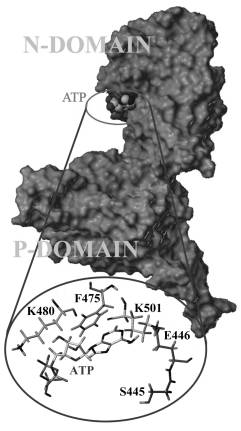
Figure
1. N and P-domain
of the H4–H5 loop of Na+/K+-ATPase
[5]. In the inset are important amino acids for the ATP-binding.
In a next step the H4–H5 loop sequence
(Leu354–Ile778) was prepared from
the sequence of the a subunit of mouse
brain Na+/K+-ATPase by polymerase chain reaction (PCR) [6]. The
purified DNA of the a subunit was amplified
by PCR with PCR primer pairs for the desired sequence and TFL polymerase. The
product was purified on agarose gel. This vector was digested with BamHI
and EcoRI and subcloned into the linearized pGEX-2T at the site location
downstream from the GST coding sequence. The ligated DNA was transformed into
competent Escherichia coli DH5a cells. The stop
codon at Lys605 was introduced using a Stratagene Quick-Change Kit
for site-directed mutagenesis. The E. coli transformants were
selected on Luria–Bertani agar containing ampicillin (50 mg/mL) and positive clones were finally sequenced. The protein was
expressed as in a previous study [7]. In a
next step a model of the N domain of mouse brain Na+/ K+-ATPase
from Leu354–Ile604 was
generated by analogy to the above mentioned model for pig kidney Na+/K+-ATPase [6]. After
docking of ATP to domain N, changes in the secondary structure were affecting
only residues close to the binding site; the overall changes were smaller than
2%. The Raman amide I band of the Raman spectra was analyzed by two different
methods for estimation of the secondary structure: least square analysis (LSA) and four
reference intensity profiles (4-RIP) with a spectrum of a solvent (Table I). Both
methods gave the same results within the maximal calculated deviation of 5 and
3%, respectively. Raman spectra can also provide additional information. Two
bands at 826 and 848 cm-1 are assigned to the Tyr (Y1 + Y16a) Fermi
dublet. The intensity ratio I848/I826 is an
indicator of the Tyr environment. We attained a value of 0.4. This indicated that
the hydroxyl groups of all three Tyr residues were donors of strong hydrogen
bonds (i.e., Tyr467, Tyr481, and Tyr535 are buried
under the accessible surface), which completely agreed with our model. The Trp
Fermi doublet intensity ratio I1360/I1340 is a
sensitive marker of the amphipathic environment of the aromatic ring. In our
case the band at about 1360 cm-1 was overlapped by the strong band
at 1342 cm-1, which made the estimation of the ratio impossible.
However, the absence of the strong band at 1360 cm-1 indicated a
hydrophilic environment for the two Trp residues. This was in good agreement
with the presence of the strong band at 755 cm-1, whose intensity is
sensitive to the amphipathic environment of the indole ring. Our
spectra thus indicated no hydrophobic interaction of Trp with neighboring
residues. The fact that our protein contains only Trp385 and Trp411 enabled us
to fit the band corresponding to the W17 normal mode for two bands with
positions at 874 and 880 cm-1. The W17 mode is sensitive to NH–H
bond donation. The band at 874 cm-1 showed
hydrogen bonding and thus corresponds to Trp385 in our
model. The Trp411, represented by the band at 880 cm-1,
does not take part in hydrogen bonding. The shift of the Phe breathing
vibration mode from 1003 to 1002 cm-1 in the difference spectrum, in
addition to ATP, was also observable. We can interpret these spectral changes
as local changes of the domain N conformation caused by the ATP binding.
Changes of the Phe band were in good agreement with the presence of Phe475 and Phe548 in the
binding site found in our model. Thus, we can say that our recombinant protein
corresponds to domain N in mouse brain Na+/K+-ATPase and docking of ATP proposed changes in the
range of 1–2% of the overall secondary structure, which is in agreement with
our experimental findings [6].
Table I. Secondary structure of domain N [6] obtained by modeling, circular dichroism (program
Varselec and CDNN) and Raman spectroscopy (method LSA and 4-RIP).
Method a-helix b-sheet b-turn other
_____________________________________________________
Model 29.6 31.2 20.4 18.8
Varseleca 25.5 27.5 – 31.5
CDNN 26.2 21.5 20.7 31.2
LSA 31.6 34.8 19.1 14.5
4-RIPa 30.3 34.9 – 34.8
_____________________________________________________
a
b-turns
are included in other structure
Site-directed mutagenesis was performed to identify
residues involved in ATP binding [8]. On the basis of our above developed model
of this loop, Ser445, Glu446, and Phe475 were proposed
to be close to the binding pocket. Replacement of Phe475 with Trp
and Glu446 with Gln profoundly reduced the binding of ATP,
whereas the substitution of Ser445 with Ala did not
affect ATP binding. Fluorescence measurements of the fuorescent analog TNP-ATP,
however, indicated that Ser445 is close to
the binding site, although it does not participate in binding. All point
mutants of our construct F475W, E446Q, and S445A were expressed as GST-fusion
proteins in E. coli and purifed. The dissociation constant of ATP to the
H4–H5-loop from Na+/K+-ATPase is about three orders of magnitude higher than
of TNP-ATP. Competition for the binding sites between ATP and TNP-ATP was used
to characterize the binding of ATP to the fusion proteins. The presence of ATP
changed signifcantly the fuorescence intensity of TNP-ATP when titrated in the
presence of all the mutants used in our study. The decrease in fuorescence
intensity in the presence of ATP indicated that some binding sites were
occupied by ATP and allowed us to calculate the dissociation constants for ATP.
We observed an increase of the value of the dissociation constant from 6.2±0.7
mM for WT to 19±2 or 14±3 mM for E446Q or F475W, respectively, suggesting an
inhibition of ATP-binding. Contrary to TNP-ATP-binding, this effect was not
observed for the mutation S445A.
In
conclusion, we show that WT protein (Leu354–Ile604) binds ATP as well as its
fluorescent analog TNP-ATP. The amino acids Phe475 and Glu446 play an important role in this
interaction. Substitution of the phenylalanine residue 475 by tryptophan and
glutamic acid 446 by glutamine affected severely the interaction with both ATP
and TNP-ATP, as indicated by a positive change in the binding energy compared
to WT. This is in good agreement, not only with our prediction from computer
modeling but also with the sequence comparison of Ca2+-ATPase
with Na+/K+-ATPase. Both methods have suggested Phe475 and Glu446 to be part of the ATP-binding site.
We can observe stacking of the aromatic ring of phenylalanine that is parallel
to the purine ring of ATP at a distance of 3 Å, in our predicted model
structure. Substitution of Ser445 by alanine did not significantly
affect ATP-binding. Nevertheless, the change in binding of the more bulky
TNP-ATP molecule indicated the residue to be in close proximity to the
ATP-binding site. In our predicted model structure, the carboxyl group of Glu446 forms a hydrogen bond over a
distance of 2.0 Å to the NH2 hydrogen donor of the adenosine moiety. The hydroxyl group of Ser445, however, is about 7.5 Å away
from the ATP molecule at the closest distance. Thus, a direct interaction seems
unlikely and our model proves to be correct in this respect. As a result we
have found beside the previously reported amino acid residues Lys480,
Lys501, Gly502 and Cys549 another four amino
acid residues, namely Glu446, Phe475, Gln482
and Phe548, completing the ATP binding pocket of Na+/K+-ATPase. Moreover,
mutation of Arg423 has also resulted in a large decrease of the ATP
binding constant. This residue, localized outside of the binding pocket, seems
to play a key role in supporting the proper structure and shape of the binding
site, probably due to formation of the hydrogen bond with Glu472
over a distance of 1.7 Å. Breaking this hydrogen bond causes probably an
instability in the stretch of amino acids containing the residues Phe475,
Lys480 or Gln482 within the binding pocket which are in
proximity to the other residues involved in ATP binding, like Lys501
or Glu446 [9]. Molecular modeling of the ATP site within the H4–H5 loop reveals that the set of these eight
amino acids residues forming the ATP recognition site is complete.
The
support by the Grants No. 204/01/0254, 204/01/100 and 309/02/1479 of the Grant Agency
of the Czech Republic and the Ministry of Education of the Czech Republic (No.
MSM113100001, No. MSM123100001) is acknowledged.
[1] P.L. Jørgensen, Kidney Int.,
29 (1986) 10–20.
[2] M. Esmann, S.J. Karlish, L. Sottrup-Jensen
and D. Marsh, Biophys. J., 70 (1994) 182–193.
[3] H. Linnertz, I. Mikšík, P. Kvasnička, E.
Bertoli, L. Mazzanti, W. Schoner and E. Amler, Eur. J. Biochem., 251
(1998) 522–527.
[4] T. Obšil, F. Mérola, A. Lewit-Bentley and E.
Amler, FEBS Lett.,426 (1998), 297–300.
[5] R. Ettrich, M. Melicherčik, J. Teisinger, O.
Ettrichová, R. Krumscheid, K. Hofbauerová., P. Kvasnička, W. Schoner and E.
Amler, Journal of Molecular Modeling, 7 (2001) 184–192.
[6] K. Hofbauerová, V. Kopecký Jr., R. Ettrich, O.
Ettrichová and E. Amler, Biopolymers,
67 (2002) 242–246.
[7] T. Obšil, K. Hofbauerová, E. Amler, J.
Teisinger, FEBS Lett.,457 (1999) 311–315.
[8] M. Kubala, K. Hofbauerová, R. Ettrich, V.
Kopecký Jr., R. Krumscheid, J. Plášek, J. Teisinger, W. Schoner and E. Amler, Biochem. Biophys. Res. Commun., 297 (2002) 154–159.
[9] M. Kubala, J. Teisinger,
R. Ettrich, K. Hofbauerová, V. Kopecký Jr., V. Baumruk, R. Krumscheid, J.
Plášek, W. Schoner and E. Amler, Biochemistry, (2003) submitted.