STRUCTURAL
BASIS OF PROTEIN METASTABILITY
Peter Flecker, Johannes Gutenberg University,
Department of Chemistry and Pharmacy, Duesbergweg 10-14, D-55128 Mainz, FRG. E-mail: flecker@mail.uni-mainz.de
All information
for the three dimensional structure of proteins and their functionality is
encrypted within their amino acid sequences. The natural amino acid sequences
of proteins have been perfected by evolution not only for their functional
structure but also for a rapid and highly directional acquisition of their
folded, fully functional state. An unambiguous distinction between these two
possibilities is important for a clear-cut interpretation of consequences of
amino acid replacements in protein engineering experiments.
The
double-headed Bowman Birk serine protease inhibitor (BBI) built up from two
triple stranded b-hairpin
domains directed against trypsin and chymotrypsin was selected as a model
protein. The double-headed arrangement of two independent subdomains
facilitates the detection of long-range irregularities transmitted from the
trypsin- into the chymotrypsin-inhibitory region used as a reference.
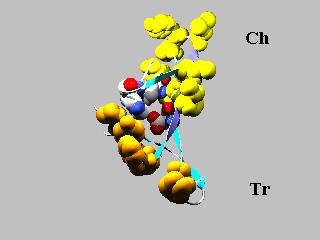
The
exposed hydrophobic patch belonging to the trypsin-inhibitory region (Tr) is
shown in ochre and that projecting out from the chymotrypsin-inhibitory region
(Ch) is shown in yellow. Residues belonging to the buried polar interior are
highlighted in CPK.
The
exposed hydrophobic patches on the protein surface and a polar protein interior
appear as structural peculiarities more reminiscent of
the kind of structural features that have been postulated to occur in partially
folded proteins rather than their folded state.
The
autonomous refolding competence of the parent protein was abolished as s result
of amino acid replacements, resulting in heterogeneous populations of
conformers greatly differing in their activity. In contrast to the autonomous
refolding competence of the parent protein, the variants require the presence
of trypsin-Sepharose as a template with complementary structure in order to
reach their fully active state. The fully active state of the variants attained
by means of this method returns to the initial mixture of conformers upon
subsequent incubation in the refolding buffer in a slow first order reaction.
Therefore, the fully active state of the variants may be regarded as local
energetic minima surrounded by high barriers of activation. The appearance of
apparently metastable state supports a kinetic reaction control for the
variants on the template although it cannot rule out a thermodynamic reaction
control. In fact, the template facilitates
folding not only kinetically, by reducing the high barrier of activation in
solution but also thermodynamically by stabilising the fully active state by
means of protein-protein-interactions. Protein metastability has also been documented
for certain proteolytic enzymes after removal of their prosequences and for the
native conformation of viral hemagglutinins. Presently, it is unknown whether
the native conformation of soybean BBI corresponds to a global energy minimum
or a metastable state on its conformational landscape. However, the inside-out
situation in BBI and the conformational changes that are induced with reducing
agents even in the absence of denaturants seem to be more in favour of the
second possibility.