MOLECULAR MODELING AS A TOOL IN MOLECULAR BIOLOGY OF MEMBRANE-BOUND RECEPTORS
R. Ettrich1, V. Vlachová2, J. Teisinger2, J. Pavlíček3, K. Bezouška3, V. Kopecký Jr.3,4 and D. Štys1
1Laboratory of High
Performance Computing, Institute of Physical Biology USB and Institute of
Landscape Ecology AS CR, University of South Bohemia,
Zámek 136, CZ-373 33 Nové Hrady, Czech Republic, email: ettrich@greentech.cz
2Institute
of Physiology, Czech Academy of Sciences, Vídeňská 1083, CZ-142 20
Prague, Czech Republic
3Department
of Biochemistry, Faculty of Sciences, Charles University, Albertov 2030, CZ-128 40 Prague 2, Czech Republic
4Institute
of Physics, Charles University, Ke Karlovu 5, CZ- 121 16 Prague 2, Czech Republic
Protein function is strongly connected to the structure. For that we need
to explore the three-dimensional structure if we want to understand the
enzymatic or structural function of a protein. Although today there are
existing several experimental techniques to determine the three-dimensional
structure of proteins, as are NMR-spectroscopy or X-ray diffraction, these
methods have their shortcomings, especially in the case of membrane proteins,
which shows the low number of known structures in the Brookhaven Protein
Database. And even if one structure is known it is often difficult to
understand the complete function, due to a lack of information with respect to
different conformational states of the membrane protein. For that reason can be
useful tool in understanding the protein function a combination of homology and
energetic modeling with vibrational spectroscopy. We confront the model already
in the process of homology modeling (restraint-based method) with data gained
by Raman and infrared spectroscopy, to have a continuous feedback. Although
these spectroscopic methods do not give such complex information, in
combination with the molecular modeling they give often enough information to
understand important functional features of the protein or help to identify
binding sites. Molecular dynamics then can explain certain dynamical features
related to the function in contrast to a static model. Thus gained models are
an important help in site-directed mutagenesis, truncation, binding-studies and
even in crystallography.
An example for a successful application of the method was achieved in the
study of the vanilloid receptor, a member of the transient receptor channel
family. Our constructed model for the first time described a folding motif for
the C-terminal tail of the channel, and according to this prediction,
truncations were constructed for electrophysiological studies. Thus the
function not only of the C-tail but also of several secondary structure
elements in it could be described in detail. Vanilloid receptor 1 (TRPV1,
formerly VR1) has been suggested to function as a multimodal signal transducer
of noxious stimuli in the mammalian somatosensory system [1]. Noxious thermal
stimuli (> 43°C), acidic pH (< 6.8) or the alkaloid irritant capsaicin
are required to open the TRPV1 channel. At room temperature and pH 7.3, TRPV1
behaves as a voltage-gated outwardly rectifying non-selective cation channel
since it can be activated strongly by depolarizing voltage steps in the absence
of any agonist [2]. Although some knowledge of the structure and function of the
TRPV channel subfamily has accumulated recently, the critical structural
domains and the mechanisms by which various external stimuli translate into
channel gating remain poorly understood. How the C-terminal domain
contributes to the conformational stability of TRPV1 channel and the extent to
which it influences its function, however, still remained to be determined. In
our recently published study [3], we demonstrate that the cytosolic C-terminal
region of TRPV1 channel contains domain(s) responsible for a steep temperature
dependence of the TRPV1 heat-evoked responses. We hypothesize that this region
is also important for regulation of the capsaicin-, low pH- and voltage-induced
channel activity. To explain and
predict the involvement of the C-terminal domain in TRPV1 channel function, the
sequence of the TRPV1 C terminus from Ala 690 to Lys 838 was used for homology
modeling. This section of the TRPV1 receptor shows a high sequence homology
(44%) to the fragile histidine triad protein FHIT, whose tertiary structure has
been solved at 1.85 Å resolution. The overall predicted structure of the
TRPV1 C-tail can be described as a general alpha+beta type and can be further
subclassed as an alpha+beta meander fold. The C-tail contains two helices, H1
and H2, and seven beta strands (Fig. 1). The strands three to seven form a
five-stranded antiparallel sheet. The antiparallel strands one and two form a
beta-hairpin across from and at an angle to the other sheet. Helix H1 packs on
one side of the five-stranded antiparallel beta-sheet, and helix H2 packs on
the same side and primarily interacts with strand three and the loop connecting
strands two and three. The template structure shows a disordered gap from
residue 107 to residue 127. Therefore, the structure of the large loop between
beta-strand 7 and the second helix was generated only from a loop database and
is not based on homology. Probably this loop is highly flexible in reality and
the shown structure must be thus taken as only one speculative possibility for
its conformation.
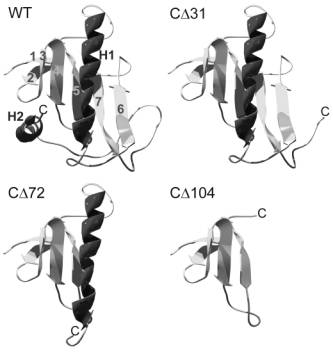
Figure 1.
Predicted structure of the
complete C terminus of TRPV1 and the truncated mutants. WT, Ribbon diagram of
the wild type C terminus (residues A690-K838). Homology modeling predicts two
alpha-helices H1, H2 and seven beta-strands 1-7. Antiparallel strands 1 and 2
form a beta-hairpin, strands 3-7 form a five-stranded antiparallel sheet. CD31, This mutant (residues A690-T807) lacks the
alpha-helix H2. CD72, In this construct (A690-C766) secondary
structural elements H2 and beta-strands 6 and 7 are missing. CD104, The predicted structure of the truncated
construct (A690-G734) consists only of beta-strands 1-5.
In the molecular model based on homology with the fragile histidine triad
protein presented here, the most distal 31 amino acid residues of the TRPV1
carboxyl terminus (Gln 808– Lys 838) correspond to the alpha helical structure
H2 and the large flexible loop connecting it with beta-sheet 7 (Fig. 1).
Removal of this region is sufficient to shift the thermal threshold for
activation from 42°C to 39°C. This structural part seems also to modulate the
sensitivity to capsaicin as the mutant CD31 exhibits increased
agonist efficacy. Deletion of the remaining short part of the connecting loop
(Arg 797–Lys 838) markedly decreased the thermal threshold (to 33°C) for
receptor activation. The large loop seems to be anchored between helix H2 and
beta-strand 7 to form itself a highly flexible structure that regulates steric
accessibility to the core beta-sheet. The effects of the deletion of the
remaining 11 loop amino acids in the mutant CD42 suggest the
importance of beta-sheets 6 and 7 in channel activation.
This view is supported also by construct CD72 which lacks
beta-strands 6 and 7 (Glu 767–Lys 838) and displays profound changes in channel
function. The thermal threshold dropped from 41.5 to 28.6°C, Q10
decreased
from 25.6 to 4.7 and the currents induced by capsaicin, pH 5, heat and voltage
decreased significantly suggesting a distinct role of these two beta-strands in
the C-terminus of TRPV1. The experimental results for the mutant CD104 presented
above indicate a disturbed multimerization of protein subunits. The helix H1 is
lost in the model of this mutant. Therefore there is a high probability that
this helix plays a role in either the tetrameric organization of the channel or
in an interaction with another receptor region, e.g. of the N-terminal. The
beta-hairpin formed by he first two antiparallel strands does not seem to
exhibit any functional role; however, it could stabilize the proper position of
the C-tail towards the membrane. In conclusion, our results provide evidence
that the structural basis of the thermal sensitivity of the TRPV1 channel
resides in the distal half of the C-terminus and that this terminal region
contributes to the regulation of chemically, thermally and voltage induced
activity of the TRPV1 channel.
As a second example, using the same technique, may serve the research
done on the melatonin receptor type 1B. Melatonin receptors are a subfamily of
G protein-coupled receptors for the pineal hormone melatonin, dubbed „the
hormone of darkness“. A molecular model of the melatonin receptor was
constructed by homology modeling from the structure of rhodopsin. The refined
model at this stage contains 194 amino acids and shows five transmembrane
segment. About 100 amino acids, which means two transmembrane segments are
still missing. At this stage it nicely shows two intracellular loops between
the first and the second and the third and fourth transmembrane segment. While
the first intracellular loop can be described as just a simple turn between the
transmembrane segments, the second from Cys 109–Ser 122 contains 14 amino
acids. This loop is an important candidate for playing a crucial role in the
channel function. It includes three tyrosines and two lysines. The three
aromatic residues are potential candidates for vibrational spectroscopy while
the lysine residues can be used for fluorescence labeling. Thus our model can
already serve as a tool for predicting proper candidates for site-directed
mutagenesis with respect to the role of the second intracellular loop. The next
step in our modeling research will be to model the complete sequence containing
also the third intracellular loop by the restraint-based method. To examine
various possible conformations of the two loops it will be necessary to perform
molecular dynamics and to verify and improve the model by means of various
spectroscopic methods. The thus confirmed model will then serve as the
workhorse for computational ligand docking as well as for experimental studies
by means of site-directed mutagenesis, truncation, binding-essays and further.
This combination of modeling and recombinant melatonin receptor provides new
tools for investigating fundamental mechanisms of melatonin action.
The last example shall show that crystallisation and X-ray diffraction
are not always able to answer important physiological questions and that
computer modeling can be a useful and even necessary addition.
CD69 is the earliest leukocyte activation antigen
playing a pivotal role in cellular signaling.
In humans, the CD69 gene is located in chromosome 12 at bands p13-p12 in
a region known as natural killer complex in association with other C-type
lectin genes that control NK cell activity. CD69 is a disulfide-linked
homodimer with two constitutively phosphorylated and variously glycosylated
chains. It belongs to the type II integral membrane protein possessing an
extracellular C-terminal protein motif related to C-type animal lectins. Since
the ligand domain of CD69 has been defined in the recent domain swap
experiments [4], we prepared constructs limited to the C-terminal portion
including residues 100–199. This construct is a minimal size
monomeric protein known to contain all amino acid residues responsible for
potential binding of calcium and carbohydrates [5].
In order to determine if CD69 binds calcium, we saturated the purified
protein with Ca2+ ions, and performed direct determinations of
calcium in the samples of protein subjected to various dialysis procedures.
More precise parameters for binding of calcium ion to CD69 were obtained from
equilibrium dialysis studies using 45Ca2+. In order to
identify the amino acids in CD69 involved in calcium binding more precisely, a
Conolly-type surface with the electrostatic potential as surface property was
generated for the published structure of this antigen. On this surface it was
possible to identify a highly negative charged region, which corresponded to a
potential calcium-binding site. Calcium was then docked into this single site
formed by aspartic acid Asp 171, and the two adjacent glutamic acids Glu 185
and Glu 187. Remarkably, the insertion of calcium into this site resulted in no
significant changes in the overall three-dimensional structure of CD69. Binding
of calcium to the wild-type protein proceeded with a dissociation constant of approximately
54 mM. In order to prove the role of the specific amino
acids in the binding of calcium, mutant proteins have been produced in which
the above amino acids, i.e. Asp 171, Glu 185 and Glu 187, have been
individually replaced by alanine. Mutation of any of the anticipated carboxyl
group of Asp 171, Glu 185, or Glu 187
resulted in considerable reduction of the affinity of calcium
binding (Kd of 0.5 mM, 0.1 mM, 0.5 mM has been estimated for the
respective mutant proteins), although the effect observed for Glu 185 has been
somewhat less profound. The double mutant with Glu 185 and Glu 187 replaced by
alanin exhibited totally no binding of calcium. The question why Ca2+
ion was not observed in the published crystal structures is explained by the
fact that the proteins used for crystallization were perhaps prepared in the
calcium-free form. We believe that the atomic structure of CD69 was then
obtained under artificial, non-physiological conditions.
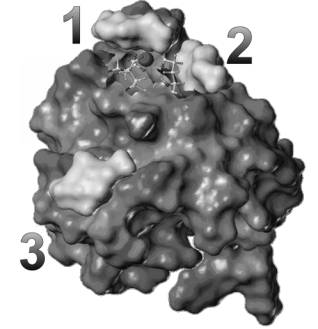
Figure 2.
Molecular details of the three GlcNAc molecules docked
into the calcium form of CD69. The potential high-affinity binding site (site 2) is
localized
close to the calcium-binding site. One low affinity binding site (site 1) is
also close to the calcium ion, while the second low affinity binding site (site
3) is in the more distal part of the molecule.
Since our recently published data [5] provide clear evidence that calcium
is an integral component of CD69 protein under physiological conditions, and
since there have been no changes in the overall structure of the protein except
in the spatial position of specific amino acid residues, we were interested to
investigate the effects of calcium binding by CD69 on the interaction with the
carbohydrate ligands. Since the most precise and correct method to address these
issues would be a direct binding assay, we decided to perform equilibrium
dialysis experiments with the labeled carbohydrates. In order to reveal the
structural details of GlcNAc binding to CD69, we have used the structure of the
calcium-ligating form of the protein, and performed molecular docking of the carbohydrate into the receptor structure
(Fig. 2). According to this model, GlcNAc bound into three sites, designated 1,
2 and 3, The identification of binding sites for N-acetylated hexosamines (3 for
GlcNAc and 2 for GalNAc) confirmed by site-directed mutagenesis (together with
the elucidation of the role of calcium in the binding process) sheds a light
onto the current controversy about the carbohydrate-binding specificity of this
protein [5]. The high-affinity binding site for GlcNAc has moreover
certain unique features not observed in other C-type animal lectins. The
unknown electron density in the Natarajan’s crystal [6] of
hexagonal, pyranose-like shape is localised directly in the position of the
carbohydrate-binding site 1 described in our work. Moreover, the arrangement
all three carbohydrate-binding sites detected here is in a good agreement with
the suggestion of Llera’s ligand-binding surface. (site 1 and 3 are directly in
this area, the site 2 is in a close proximity) [7]. Altogether, identification
of binding sites for calcium and for monosaccharides now open the way for
searching of complex oligosaccharides as the potential physiological ligands
for CD69.
The support by the Grant Nos. 309/02/1479, 305/03/0802 and 203/01/1018 of the Grant Agency of the Czech Republic and the Ministry of Education of the Czech Republic (No. MSM113100001, No. MSM123100001, No. MSM113200001) is acknowledged.
[1] Caterina M.J.,
Schumacher M.A., Tominaga M., Rosen T.A., Levine J.D., Julius D., Nature 389 (1997), 816–824.
[2] Vlachova V.,
Susankova K., Lyfenko A., Kuffler D.P., Vyklicky L., Psychiatrie 6 (2002), 6–13.
[3] Vlachova V., Teisinger J., Susankova K., Lyfenko
A., Ettrich R., Vyklicky L., Journal of Neuroscience 23 (2003), in
press.
[4] Sancho, D., Santis, A.G., Alonso-Lebrero, J.L., Viedma, F.,
Tejedor, R., and Sánchez-Madrid, F, J.Immunol. 165
(2000), 3868–3875.
[5] Pavlicek J., Sopko B., Ettrich R., Kopecký Jr. V., Baumruk V., Man P., Havlíčková
K., Vrbacký M., Křen V., Pospíšil M. and Bezouška K., Biochemistry (2003)
submitted.
[6] Natarajan, S., Sawicki, M.W., Margulies, D.H., and
Mariuzza, R.A., Biochemistry 39 (2000), 14779–14786.
[7] Llera, A.S., Viedma, F.,
Sanchez-Madrid, F., and Tormo, J., J.
Biol. Chem. 276 (2001), 7312–7319.