A program for automatic checking of crystal structure solution results based on comparison with DFT calculation results
F. Fňukal
University of Chemistry and Technology, Prague, Technická 5, 166 28 Praha 6 – Dejvice
fnukalf@vscht.cz
Introduction
Crystal structure verification based on the
comparison with DFT calculation results was already introduced circa 20 years
ago [1, 2]. However, only the advancement in computing technology as well as
the development in the area of DFT functionals made it possible to perform such
calculations on complex organic molecular crystals. Our aim is to develop a
program capable of mediating DFT calculations and analysing the results. There
already exist commercial pieces of software offering such capabilities, they
are however typically fairly expensive. Our aim is therefore also to present a
freely available variant of such software.
Crystal structure verification using dispersion-corrected DFT
A DFT calculation uses an experimental structure as
an input. During the calculation, the atomic positions and optionally also the
cell parameters are optimized in a way that the energy minimum is achieved. The
output of a DFT calculation is another structure with a geometry more or less
different from the geometry of the experimental structure. The input and output
structures can then be compared based on certain selected criteria. These
criteria should indicate serious discrepancies in the two structure geometries.
Our implementation – the program checkCIF-DFT
To facilitate performing DFT calculations on
crystal structures we developed a program to which we gave the name
checkCIF-DFT. An inspiration to us was the web application checkCIF/PLATON [3],
which offers consistency and validity checking for experimental crystal
structures based on crystallographic diffraction criteria. Our program
intrinsically utilizes 3 different DFT programs: Quantum ESPRESSO [4],
CASTEP [5] and Orca [6]. Besides that, the molecular mechanics program GULP [7]
is also utilized. Our program provides a graphical interface and serves as a
mediator between the user and computational programs. Our program can read and
visualize data from a CIF file, prepare input files for computational programs,
monitor the progress of a calculation and finally, after a calculation has
finished, it can analyse the calculation results and point out serious issues.

Figure 1. Main window of the program checkCIF-DFT.
Input and output structures comparison
To compare the experimental crystal structure and
the DFT output crystal structure, it is absolutely essential to choose
comparison descriptors that are sufficiently indicative and can therefore
reflect serious discrepancies in the compared structures. In our work, we
originally used solely the descriptor RMSCD developed by other authors [2].
However, as the authors of RMSCD stated themselves, this descriptor doesn’t
reflect serious issues well enough. For that reason, we decided to include
other descriptors. Among the newly implemented descriptors are relative
difference in cell volumes, maximal difference in bond lengths, maximal
difference in bond angles and others. In our testing so far, we discovered that
the tested problematic structures reliably exhibit a serious disagreement in at
least one of the used descriptors.
Practical uses of DFT calculation results
DFT calculations can be used for routine
verification of experimental crystal structure solutions. Some experimental
results may be affected by serious errors due to bad quality of the crystalline
sample or other factors. For that reason, a DFT calculation can be useful to
assess the trustworthiness of the experimentally obtained data.
Crystal structure prediction represents another field of use for DFT
calculations. In such computational experiment a large set of possible crystal
structure geometries is generated using lower-level methods (e.g. molecular
mechanics). These structures are then refined using the DFT method. The refined
set of structures can then be sorted based on the lattice energy, which should
reflect the stability of each structure in the set.
Apart from the two examples mentioned above, DFT calculations also find great
use in powder diffraction crystal structure solutions. While solving powder diffraction
data, a DFT calculation can be used as an intermediate step to achieve a better
level of refinement.
DFT method testing
In our work, we’ve conducted a series of testing calculations
to assess how well the DFT method would fare in indicating seriously erroneous
crystal structure solutions. In our testing, we chose a set of 5 structures
that are known to be fraudulent [8] and a set of 5 structures solved by neutron
diffraction experiments, which we deemed to be the most precise and reliable
method of determining the crystal structure. For this test we used the CASTEP
computational module utilizing the rSCAN functional and MBD dispersion
correction. When analysing the results, we concluded that the DFT method
together with our improved descriptor system was able to detect that the
fraudulent structures were erroneous (Fig. 2).
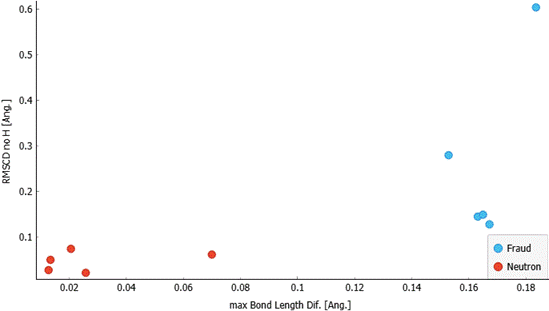
Figure 2. Scatterplot of RMSCD excluding hydrogen atoms against maximal bond length difference for a set of structures solved using neutron diffraction data (red) and a set of structures that are known to be fraudulent (blue).
We also used the DFT method in a crystal structure prediction computational experiment. DFT calculations were performed on a set of 100 trial structures with the code XXXI from the 7th CSP Blind Test [9]. In this test we used the Quantum ESPRESSO computational module utilizing the PBE functional and D3 dispersion correction. Using our method, we were able to capture the 3 experimentally observed polymorphs among the first 8 structures with lowest calculated lattice energies (Tab. 1). The DFT method is however known to not yield perfect results in this type of crystal structure prediction experiment, mainly due to difficulties in describing thermal effects.
Table 1. Best 10 trial structures of the compound XXXI from the 7th CSP Blind Test as calculated by the DFT method.
|
Rank |
Structure code |
Relative energy [kJ/mol] |
Experimental rank |
|
1. |
XXXI_structure_59 |
0.0000 |
- |
|
2. |
XXXI_structure_98 |
0.7963 |
1. |
|
3. |
XXXI_structure_1 |
2.0061 |
2. |
|
4. |
XXXI_structure_17 |
2.5395 |
- |
|
5. |
XXXI_structure_57 |
2.9341 |
- |
|
6. |
XXXI_structure_34 |
3.0854 |
- |
|
7. |
XXXI_structure_11 |
3.1684 |
- |
|
8. |
XXXI_structure_25 |
3.2349 |
3. |
|
9. |
XXXI_structure_70 |
3.4779 |
- |
|
10. |
XXXI_structure_20 |
4.1705 |
- |
Conclusions
In our work, we discovered that we were able to
detect fraudulent crystal structures using DFT calculations together with an
improved system of structure comparison descriptors. The most useful comparison
descriptors have shown to be the maximal bond length difference and maximal
bond angle difference.
We developed and tested a freely available program that is capable of mediating
DFT calculations and analysing the results. This program may help
crystallographers in assessing the trustworthiness of crystal structure
solutions.
1. J. Streek, M. A. Neumann, Acta .Cryst., B66, (2010), 544.
2. J. Streek, M. A. Neumann, Acta .Cryst., B70, (2014), 1020.
3. (IUCr) IUCr Journals - checkCIF FAQ. https://journals.iucr.org/services/cif/checking/checkfaq.html#what (accessed May 10, 2024).
4. Paolo Giannozzi et al, J. Phys.: Condens. Matter, 21, (2009), 395502
5. Materials Studio 2023 - CASTEP. https://www.tcm.phy.cam.ac.uk/castep/documentation/WebHelp/CASTEP.html (accessed May 10, 2024).
6. F. Neese, Wiley Interdisciplinary Reviews: Computational Molecular Science, 2, (2012), 73
7. J. Gale, J. Chem. Soc., Faraday Trans., 93, (1997), 629
8. The Lancet, 375, (2010), 94
9. The 7th CSP Blind Test | CCDC. https://www.ccdc.cam.ac.uk/community/ccdc-for-the-community/partnerships-and-initiatives/csp-blind-test/7th-csp-blind-test/ (accessed May 10, 2024).