The potential of dispersion-corrected density functional theory calculations to verify CSD database content
M. Hušák
1University of Chemistry and
Technology, Prague
husakm@vscht.cz
Introduction
The idea to verify crystal structure by comparison with DFT calculation results was introduced already 20 years ago [1,2]. Due to advances in the computation technologies, DFT functionals development as well as bit problematic results of the original work we had chosen to refresh the whole idea. The target of our work is to develop this methodology and to test whatever verification of the complete CSD content by this method is eventually possible.
Improvement of the methods
The original verification method [1,2] was primary based on the experimental and DFT results comparison by Cartesian displacement (RMSCD) descriptor only. RMSCD is de-facto a RMSD modified to be able to compare atomic positions in different unit cells. It was already mentioned [1] this descriptor was not able to clearly separate totally artificial fraud structures from correctly solved one. We had tested several other descriptors to separate problematic structures from correct ones. Our test indicates the maximal difference in bond distance and maximal difference in bond angle can better indicate problematic structures than RMSCD, see Fig. 1. The artificial fraud structures are clearly separated from correct results.
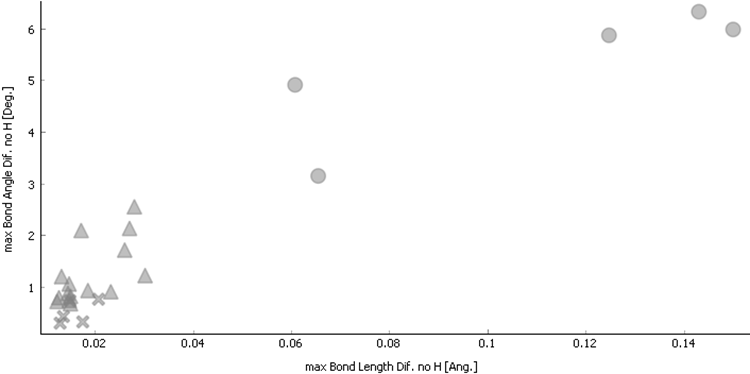
Figure 1. Maximal bond length difference versus maximal bond angle difference (hydrogen atoms excluded). Ï - correct neutron structures, Δ - correct X-ray structures, ™ - artificial fraud structures
Another improvement of the method is the use of modern meta-GGA functional (r2SCAN) with up-to-date dispersion correction (MBD) instead of the PBE functional and first generation of the Grimme dispersion correction as used in the original work. This method of energy calculation leads to more realistic atom positions and lattice parameters than the original one. The whole methodology was implemented in a form of checkCIF-DFT software which is a subject of another presentation.
Test on 100 semi-randomly selected structures from CSD
In the first tests we had extracted from CSD 194017 structures by following criteria: published after 2013, non-disordered pure organic structures, no errors detected by CSD, solved from single crystal. We had used only pure organic structures because metal presence can generate problems related to open shell and not clear spin state which is hard to handle automatic for DFT calculation. From the mentioned structures we had selected in a semi-random way 100 one (with one addition criteria - original diffraction data must be deposited). For 30 structures it was impossible to perform the verification - see Tab. 1 for reasons.
|
Issue description |
Number of structures |
|
Incorrect space group |
1 |
|
Missing disordered on nonsense hydrogen atoms positions. |
4 |
|
Voids indicating missing solvents (Solvent Area > 40 Å3) |
12 |
|
To big structures (performance issues) |
2 |
|
Not converging after 100 optimization steps |
7 |
|
Not fitting in used computer memory |
1 |
|
Duplicated atoms generated over symmetry |
3 |
|
Correctly calculated structures |
70 |
Table 1. Issues detected during the CSD 100
structures test
The
verification process was fully finished for 70 structures from the test set.
The maximal bond and angle difference descriptor visualisation graph is on the
Fig 2.
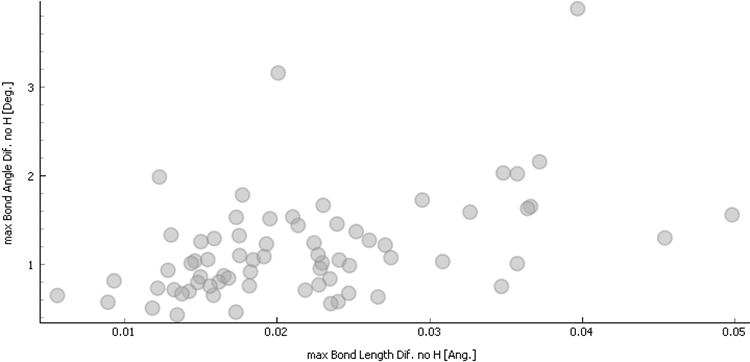
Figure 2. Maximal bond length difference versus maximal bond angle difference (hydrogen atoms excluded) for the 70 fully processed structures.
Pre filtering test on the bigger structures sample
Based on the issues with the 100 structures sample, we had tried to run some simpler test on the whole 194017 set. Eventual computational non-expensive pre-filtering can help to save computational resources as well. The inspiration of the test was the PLATON/checkCIF code.
The first test was a test for correct space group determination. The test checks whatever there exist a higher symmetry able to describe the structure or whatever the structure is not described by super cell. The test was done by the help of Spglib[3] library in a similar way as it is done by ADDSYM code in PLATON. An issue was detected for 622 structures (0.32%).
Another test was done for Solvent Volume presence calculation in the unit cell. The Solvent Volume was calculated as suggested in the BYPASS article [4] so it corresponds exactly to values calculated by PLATON/checkCIF. 20 057 structures (10.34 %) with Solvent Area > 40 Å3 was detected.
The last test was done by checking the correspondence between chemical_formula_sum information in the CIF file and formula generated from tom coordinates. A disagreement was found for 45 974 structures (23.70%). In most often cases the disagreement is the result of generating more than asymmetric unit cell atoms by CSD ConQuest to get a complete molecule. Unfortunately, this data cannot be used directly for DFT calculation without pre-processing. In a lot of cases the results indicate missing atoms in the structure as well.
Conclusions
DFT method can reliably detect issues with incorrectly determined structures. Unfortunately, its use is limited by computational resources, presence of metals in the structure and structure disorder occurrence. Only a sub-set of CSD can be checked by this methodology due to multiple issues with deposited structures. Even on a small 70 structures sample from CSD, structures with symptoms similar to fraud structures (compare Fig 1 and Fig 2) can be detected. A test made on large sample by the use of supercomputer, or a test done by alternative methods (ML force field) is required for more conclusions.