Application of DFT calculations for crystal structure verification of phases from salt-cocrystal continuum area
S. Chalupná (maiden name Šajbanová), M. Hušák
Department of Solid State Chemistry, University of Chemistry and Technology, Prague, Technická 5, Praha 6, 166 28, Czech Republic
sajbanos@vscht.cz
Introductions
Pharmaceutical solid forms such as salts and cocrystals play a crucial role in pharmaceutical applications. The difference between salt and cocrystal is given only by the position of single hydrogen [1], making it essential to develop precise techniques for identifying this position. Differentiation between salt and cocrystal compounds holds significant importance within the pharmaceutical industry, both for regulatory purposes and overall quality control. We are developing a computational method for salt cocrystal differentiation based on DFT (density functional theory) energy calculation. We had already partially tested this method [2]. In this work we confirmed that we can correctly differentiate salt and cocrystal when the H-bond is not extremely strong (reliability rule set by us for H-bond distances longer than 2.613 Å, O–H···N case). Based on the conclusions from the publication we had made a few improvements in our present work: the number of tested structures increased from 95 to 404 and for the main screening the rSCAN functional was used instead of PBE one. Our DFT method optimizes an artificially constructed wrong structure (hydrogen atom placed in salt position near the potential acceptor for cocrystals and vice versa cocrystal position with hydrogen atom placed near the potential donor of the salts). The verification of the method was done based on comparison of the results with an experimentally confirmed correct hydrogen position. 16 cocrystals from the studied set were identified as salt in disagreement with experimental data. These problematic structures were investigated more deeply. We reproduced crystallization and data collection using single-crystal X-ray diffraction (SCXRD) for 7 of them. Complete experimental data were available for 2 problematic structures from the original authors and data re-interpretation was possible. To get the best possible hydrogen positions, we had used for refinement the HAR method as implemented in Olex2 software and NoSpherA2 [3,4,5,6]. We also evaluated whether in these problematic cases more advanced functionals (r2SCAN, PBE0, PBE50) could provide results consistent with experimental data.
Methods
The DFT calculations were performed using CASTEP code [7]. Since the cell parameters were assumed to be accurately obtained experimentally, we solely performed only optimization of atomic positions. We had used rSCAN functional with MBD dispersion correction and automatic fine basis precision [8,9]. The data were prepared in checkCIF-DFT software [10]. The optimization was always performed from both artificial salt and cocrystal starting models. Computation was performed on Karolina supercomputer at TU Ostrava, Czech Republic.
For cases where we have crystallized the structure or reinterpreted the data of the original authors, we used HAR method as implemented in Olex2 software and NoSpherA2 module [3,4,5,6]. For the wavefunction calculation we had used def2-TZVP localized base, r2SCAN functional and Orca 5.0 software [11]. The refinement was in all cases done by two methods. The first method was based on refinement of the problematic hydrogen in single position. The second method was based on refinement of this hydrogen in two positions as disordered one. The donor and acceptor distances to the hydrogen were in the second case restrained to the value 0.95 Å with weight corresponding to 0.01 Å esd. For final CIF deposition, the refinement results based on the first method were used only because we believe the disorder model does not correctly reflect the real state of the phases.
Results
We had confirmed a correct cocrystal structure determination in 301 cases. For 87 structures we had identified that the phase determination is suspicious, and the structures probably create a salt-cocrystal continuous phase. This phases behaviour will be described in separated study.
Table 1. Results of calculation on 404 structures from the zone with -1 ≤ ΔpKa ≤ 4 (RSCAN fine + MBD)
|
Pure cocrystal |
Pure salt |
Salt-cocrystal continuum phase |
|
301 |
16 |
87 |
From the 16 phases exhibiting consistent salt behaviour by our methodology, we experimentally proved that 2 are true salts (OGEPIA, GIPQAX). We believe that 3 others (ODOHIZ, CITSAZ10, VODCOH) were incorrectly solved by the original authors, and our DFT method using rSCAN functional correctly identified these as salts. For the 4 structures that violate the reliability rule we established in the previous article (DFT method can correctly differentiate salt and cocrystal when the H-bond is not extremely strong; H-bond distances longer than 2.613 Å, O–H···N case), we confirmed the DFT methodology based on rSCAN functional works correctly and the problem is with the experimental structure determination. In 9 cases we confirmed that the DFT method based only on rSCAN functional is not reliable and unsuitable for cocrystal/salt distinguishing with strong H-bond. However, advanced functionals (r2SCAN, PBE0, PBE50) can correct these discrepancies in some cases. For future prediction we suggest for the salt-cocrystal differentiation the r2SCAN functional which provides correct results for O–H···N bonds longer than 2.554 Å, compared to our previous 2.613 Å limit. The computational cost of r2SCAN is comparable to rSCAN, making it suitable for large-scale screening.
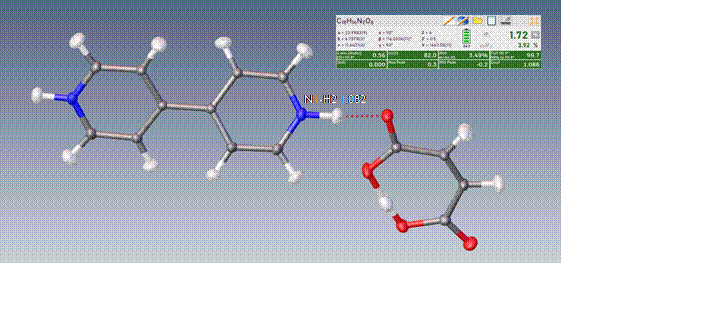
Figure 1. Structure of 4,4'bipyridine and maleic acid (GIPQAX) in Olex2 refined by HAR method with hydrogen atoms treated as anisotropic. The structure was originally experimentally solved incorrectly as cocrystal.
1. Aitipamula, S.; Banerjee, R.; Bansal, A. K.; Biradha, K.; Cheney, M. L.; Choudhury, A. R.; Desiraju, G. R.; Dikundwar, A. G.; Dubey, R.; Duggirala, N.; Ghogale, P. P.; Ghosh, S.; Goswami, P. K.; Goud, N. R.; Jetti, R. K. R.; Karpinski, P.; Kaushik, P.; Kumar, D.; Kumar, V.; Moulton, B.; Mukherjee, A.; Mukherjee, G.; Myerson, A. S.; Puri, V.; Ramanan, A.; Rajamannar, T.; Reddy, C. M.; Rodriguez-Hornedo, N.; Rogers, R. D.; Row, T. N. G.; Sanphui, P.; Shan, N.; Shete, G.; Singh, A.; Sun, C. Q. C.; Swift, J. A.; Thaimattam, R.; Thakur, T. S.; Thaper, R. K.; Thomas, S. P.; Tothadi, S.; Vangala, V. R.; Vishweshwar, P.; Weyna, D. R.; Zaworotko, M. J., Polymorphs, Salts and Cocrystals: What's in a Name? (vol 12, pg 2147, 2012). Cryst. Growth Des. 2012, 12 (8), 4290-4291.
2. Hušák, M.; Šajbanová, S.; Klimeš, J.; Jegorov, A. (2022) Acta Crystallographica Section B. 78, 5, 781 – 788.
3. Jayatilaka & Dittrich., Acta Cryst. 2008, A64, 383.
4. Capelli et al., IUCrJ. 2014, 1, 361.
5. Kleemiss et al., Chem. Sci. 2020, DOI: 10.1039/D0SC05526C.
6. Midgley et al., Acta Cryst. 2021, A77, 519-533.
7. Clark, S. J. , Segall, M. D. , Pickard, C. J. , Hasnip, P. J. , Probert, M. J. , Refson, K & Payne M. C. (2005) Zeitschrift fuer Kristallographie 220(5-6), 567-570.
8. Bartók, A. P. & Yates, J. R. (2019). J. Chem. Phys. 150, 161101-1-161101-5.
9. Hermann, J., & Tkatchenko, A. (2018) J. Chem. Theory Comput. 14(3), 1361–1369. doi:10.1021/acs.jctc.7b01172.
10. Fňukal, F Automatická kontrola výsledků řešení krystalové struktury na základě srovnání s výsledky DFT výpočtu., Vysoká škola chemicko-technologická v Praze, 6.6. 2024.
11. Neese, F. (2022). Software update: The ORCA program system—Version 5.0. Wiley Interdisciplinary Reviews. Computational Molecular Science, 12(5). https://doi.org/10.1002/wcms.1606.
This work was supported by the grant of Specific university research—grant No. A2_FCHT_2024_054.