Structural insight into antibiotic-inactivating enzyme from Stenotrophomonas maltophilia
M. Malý1,2, P. Kolenko1,2, J. Dušková1, T. Kovaľ1, T. Skálová1, M. Trundová1, J. Stránský1, L. Švecová1, K. Adámková1,3, B. Husťáková1,3, J. Dohnálek1
1Institute of Biotechnology of the Czech Academy of Sciences, Biocev, Průmyslová 595, Vestec
2Czech Technical University in Prague, Faculty of Nuclear Sciences and Physical Engineering, Břehová 7, Prague
3University of Chemical and Technology Prague, Department of Biochemistry and Microbiology, Technická 5, Prague
martin.maly@ibt.cas.cz
Stenotrophomonas maltophilia is an opportunistic bacterial pathogen responsible for a serious number of infections globally. It exhibits broad antibiotic resistance that has been further extended via the acquisition of antibiotic-resistance genes and mutations [1]. We carried out a bioinformatic analysis of its sequenced genomes to investigate not yet characterised antibiotic-inactivating enzymes.
Several chosen proteins were expressed in Escherichia coli strain Lemo21 (DE3) and purified using Ni-NTA and size-exclusion chromatography. Their proposed function – enzymatic modification of antibiotics – was inspected with an activity assay. The enzyme with the confirmed activity was successfully crystallized and diffraction patterns were collected. The data exhibited serious anisotropy: a resolution cutoff determined in Aimless [2], according to the criterion of CC1/2 > 0.30, varied from 2.43 Å to 1.92 Å in different reciprocal space directions. Thus, the data were corrected with STARANISO [3]. After the solution of the phase problem in MoRDa [4], the model was refined in REFMAC5 [5]. The choice of the anisotropic high-resolution diffraction limit (1.88 Å) was confirmed with paired refinement in PAIREF [6].
The solved X-ray crystal structure reveals an atomic arrangement of the putative substrate-binding pocket that allows further structural analysis (in silico or in vitro) of complexes with potential inhibitors or antibiotic substrates. The overall fold is very close to the tetracycline destructases [7] or the reductase involved in the abyssomicin biosynthesis pathway [8]. However, the putative active site differs significantly. Our study leads to a better understanding of the involvement of this enzyme in the antibiotic resistance and could contribute to the development of new strategies of antibiotic therapies. Remarkably, the solved structure is composed of a homodimer linked with two disulfides. Nevertheless, further investigation using small-angle X-ray scattering, mass spectrometry and dynamic light scattering showed that the protein is monomer in solution.
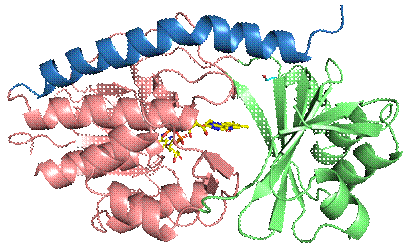
Figure 1. Overall structure of the antibiotic-inactivating enzyme from Stenotrophomonas maltophilia in secondary structure representation. Flavin adenine dinucleotide (FAD) is shown in stick representation in yellow. The substrate-binding domain is coloured in green, the FAD‑binding domain in pink and the C-terminal helix in blue.
1. T. Gil-Gil, J. L. Martínez, P. Blanco, Expert Review of Anti-infective Therapy, 18, (2020), pp. 335-347.
2. P .R. Evans, G. N. Murshudov, Acta Cryst., D69, (2013), pp. 1204-1214.
3. I. J. Tickle, C. Flensburg, P. Keller, W. Paciorek, A. Sharff, C. Vonrhein, G. Bricogne. (2018). STARANISO (http://staraniso.globalphasing.org/cgi-bin/staraniso.cgi). Cambridge, United Kingdom: Global Phasing Ltd.
4. A. Vagin, A. Lebedev, Acta Cryst., A71, (2015), s19.
5. G. N. Murshudov, P. Skubak, A. A. Lebedev, N. S. Pannu, R. A. Steiner, R. A. Nicholls, M. D. Winn, F. Long, A. A. Vagin, Acta Cryst., D67, (2011), pp. 355-367.
6. M. Malý, K. Diederichs, J. Dohnálek, P. Kolenko, IUCrJ, 7, (2020), 681-692.
7. J. L. Markley, T. A. Wencewicz, Frontiers in microbiology, 9, (2018), 1058.
8. J. A. Clinger, X. Wang, W. Cai, Y. Zhu, M. D. Miller, C. G. Zhan, S. G. Van Lanen, J. S. Thorson, G. N. Phillips Jr., Proteins. 89, (2021), pp. 132-137.
This work was supported by the MEYS CR (projects CAAS – CZ.02.1.01/0.0/0.0/16_019/0000778, BIOCEV – CZ.1.05/1.1.00/02.0109, and ELIBIO – CZ.02.1.01/0.0/0.0/15_003/0000447) from the ERDF fund; by the Czech Academy of Sciences (86652036); by the GA CTU in Prague (SGS22/114/OHK4/2T/14); by the Czech Science Foundation (20-12109S); and from the grant of Specific university research (A1_FPBT_2021_003). We acknowledge CMS-Biocev (Biophysical techniques, Crystallization, Diffraction, Structural mass spectrometry) supported by MEYS CR (LM2015043 and LM2018127).