Structural studies of purine nucleoside phosphorylase inhibitors
S. Djukic1, J. Skácel1, J. Brynda1,2,
P. Pachl1, T. Vučková1, M. Fábry2, J. Snášel1,
M. Rumlová3, T. Bílek1,3, J. Voldřich1,3, H. Mertlíková-Kaiserová1,
Z. Janeba1, P. Řezáčová1,2
1Institute of Organic Chemistry and Biochemistry, AS CR, Prague 6, Czech Republic
2Institute of Molecular Genetics, AS CR, Prague 4, Czech Republic
3University of Chemical Technology, Prague 6, Czech Republic
Stefan.dukic@uochb.cas.cz
Nucleic acid synthesis and degradation are ongoing metabolic processes in most cells. The degradative processes lead to the release of free purines and the salvage pathway exists to recover them efficiently in a useful form. Purine nucleoside phosphorylase (PNP) represents one of the key enzymes of the purine salvage pathway, which is considerably more energy-efficient than de novo pathway. It hydrolyses ribose from inosine and guanosine in the presence of an inorganic phosphate, producing hypoxanthine and guanin, which can then be recycled trough the salvage pathway or be further degraded to uric acid.
Human PNP activity is increased in T-cell leukemia, breast and colon cancer and during autoimmune diseases. It is also found to cleave antiviral drugs and to play a role in immune response, which leads to transplant-organ rejection. PNP is conserved in most organisms, and for lot of parasitic organisms like Mycobacterium tuberculosis (tuberculosis causing agent) and Plasmodium falciparum (malaria causing agent) it more favorable, if not the only way to obtain purines. Thus, human PNP, as well as parasitic PNPs, have been established as prospective targets for drug design with several PNP inhibitors recently entered human clinical trials. Due to structural similarities between PNPs in different organisms, it is a serious challenge to design selective inhibitors making structural studies of PNPs an important task.
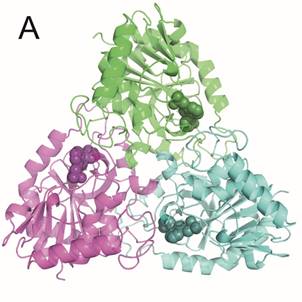
Human PNP is a functional homotrimer. Parasitic PNPs are either trimers (MtbPNP) or hexamers (PfPNP). Each subunit has an active site which is formed mostly by the parent subunit with one or few participating residues belonging to neighboring subunit.
We are using X-ray crystallography in structure-based drug design of novel acyclic nucleotide analogues. Our goal is to design inhibitors with high affinity towards hPNP as well as MtbPNP and PfPNP.
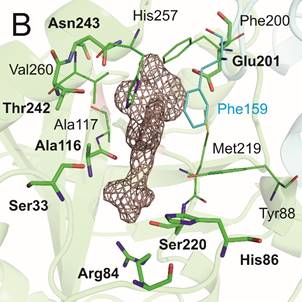
Enzymes were prepared by heterologous expression in E. coli and purified in high yields and purity necessary for crystallographic studies. Crystallization conditions for all three enzymes were identified through wide screening and optimization. Selected inhibitors with affinity in nanomolar range were successfully co-crystallized with hPNP and MtbPNP, diffraction data have been collected on BL14.1 at the BESSY II electron storage ring operated by the Helmholtz-Zentrum Berlin and crystal structures were determined at high resolution (Figure1).
The knowledge of binding properties of these inhibitors will provide us crucial information which will be used to further optimize affinity and selectivity of new generation of PNP inhibitors.