Predikování krystalových struktur
Jan Drahokoupil
Institute of Physics of the
Czech Academy of Sciences, Na Slovance 1999/2
Prague 8 182 21 Czech Republic
draho@fzu.cz
.Keywords: crystal structure prediction, DFT, molecular mechanics, global optimization
Predikování krystalových struktur látek, založené pouze na znalosti
základních stavebních jednotek (atomů či molekul), bylo cílem fyziků od
padesátých let. Ještě v roce 1988 popsal John Maddox,
jako editor časopisu Nature, stav oboru touto
výstižnou větou: “One of
the continuing scandals in the physical sciences is that it
remains in general impossible to predict the structure of
oven the simplest crystalline solids from a knowledge
of their chemical composition.”[1]. Jako taková se predikce krystalových struktur začíná
formovat okolo přelomu tisíciletí, kdy byl také vyhlášen první "blind
test" [2]. Úspěchy tohoto oboru byli podmíněny prudkým rozvojem a nyní i
cenovou dostupností výpočetní techniky. Jde o hledání kompromisů mezi přesností
popisu meziatomových sil a jejich výpočetní náročností. Problém nalezení
globálního minima se řeší chytrými algoritmy, jako jsou evoluční algoritmy,
různé metody simulovaného žíhání či relativně nové mechanizmy
jako „Particle swarm optimization“ (optimalizace hejnem částic) [3]. Pro
porovnání energií mezi jednotlivými kandidáty se využívá nejčastěji DFT či
molekulární mechanika (MM).
Významnou událostí jsou tzv. blind testy pořádané organizací CCDC.
Účastníci dostanou plošný nákres několika málo organických molekul a jejich
cílem je správně předpovědět krystalovou strukturu daných molekul. Tato
struktura je už vyřešena pomocí klasických difrakčních dat, ale ještě nebyla
veřejně publikována. První blind test běžel v roce 1999 [2] a pár let
později byl zas otevřen další [4-8]. Poslední, sedmý, blind test byl spuštěn letos
v říjnu [9]. Cílem této události je získat aktuální přehled o úspěšnosti a
problémech daného oboru. Od šestého blind testu účastníci poslali seřazený list
100 možných předpověděných krystalových struktur pro danou molekulu,
v předešlých ročnících to byli pouze 3. Pro zajímavost, v šestém
blind testu jeden účastník předpověděl správnou krystalovou strukturu jedné
molekuly s využitím pouze 26 procesorových hodin a naopak tým, který na
danou strukturu použil 30 000 000 procesorových hodin, byl neúspěšný.
Nalezení vhodného kandidáta na pravděpodobnou krystalovou strukturu je jako
hledání jehly v kupce sena. Počet možných kombinací je veliký a na cestě
k úspěšnému nalezení globálního minima stojí řada lokálních minim.
S úspěchem se využívají algoritmy globální optimalizace. Kromě,
krystalografům dobře známých, jako je simulované žíhání či paralelní
temperování, které se využívají při řešení struktury v přímém prostoru, se
používá řada dalších přístupů. Některé z nich se inspirovali přírodou,
jako jsou evoluční algoritmy [10], či optimalizace houfem částic [11] nebo „ant
colony optimization“ [12],
jiné vycházejí z pravděpodobného tvaru potenciální plochy jako metadynamika [13], „basin hopping“ [14], „minima hopping“
[15], a další.
Z komerčních programů můžeme jmenovat například Materials
studio či Grace. Pro akademické uživatele jsou zdarma dostupné například tyto
programy:
XtalOPt [16] – volně stažitelný, pro platformy Windows, Linux, Mac, používá
evoluční algoritmus, pro vyhodnocení energií externě volá Gulp
(MM), Vasp (DFT), Quantum Esspreso (DFT), Castep (DFT) nebo
Siesta (DFT).
Calypso [17] – po registraci, Linux, používá optimalizaci houfem částic,
externě volá Gulp (MM), Vasp
(DFT), Quantum Esspreso (DFT),
Castep (DFT), Siesta (DFT), nebo CP2K (DFT+MM)
Uspex [18] po registraci, Linux, používá evoluční algoritmus, ale umožňuje také optimalizaci
hejnem částic, metadynamikou nebo s využitím náhodného
vzorkování, externě volá Lammps (MM), Gulp (MM), Vasp (DFT), Quantum Esspreso (DFT), Castep (DFT), Siesta (DFT) nebo CP2K (DFT+MM).
Základní postup při predikování krystalové struktury je naznačen na Obr. 1.
Pro látky pro jejichž popis nevyužíváme molekuly, ale jen samostatné atomy, začínáme
v pravém sloupci od části „balení“. Pro molekulární látky začínáme tzv.
konformační analýzou, kdy se pomocí změny volných paramterů
dané molekuly snažíme najít její tvar (tvary) s co nejmenší energií. Je
sice pravděpodobné, že v krystalu tvar molekuly nebude úplně odpovídat
tvaru molekuly s nejmenší energií ve volném prostoru, ale zároveň síly
mezi molekulami nejsou v krystalických látkách veliké a dá se najít určité
množství tvarů, které se od „ideálního“ tvaru liší jen o určité množství
energie. Pro výběr vhodných kandidátů pro krystalickou formu můžeme použít
několik nástrojů. Co je pro nás důležité, je jejich výpočetní náročnost. Geometrická
optimalizace je náročnější než jen výpočet energie pro daný tvar a ten může být
zase výpočetně náročnější než třeba porovnávání torzních úhlů. Řekněme, že pro naší
molekulu jsme nagenerovali velký počet náhodných konformerů, jejich počet, dále můžeme zredukovat na základě
podobnosti torzních úhlu. Pro tento menší počet můžeme spočítat energii pomocí
MM. Pro několik konformerů s nejmenší energíí můžeme použít výpočet energie pomocí náročnějších,
ale přesnějších výpočtů („hrubé“ DFT). Na základě hodnot energií získaných
pomocí MM a hrubého DFT můžeme stanovit nepřesnost určení energii MM oproti
hrubé DFT a stanovit okno propustnosti konformerů do
dalšího kola. Tím může být jemnější (přesnější) nastavení DFT nebo se můžeme
pustit do geometrické optimalizace. Ta může být opět provedena v několika
kolech. Po jejím provedení můžeme opět přejít ke klastrování a spojit pod jeden
konformery s velmi podobným tvarem. Opět
provedeme energetické hodnocení a můžeme se pustit do umísťování
nejpravděpodobnějších konformerů do krystalu.
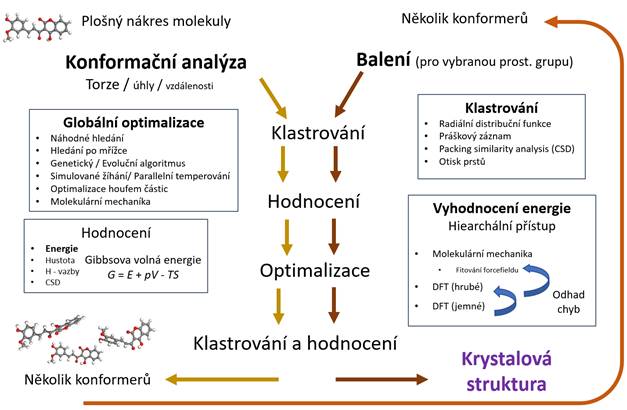
Obrázek 1. Schématické znázornění procesu
hledání/předpovídání krystalové struktury organické molekuly.
Při umísťování konformerů do krystalu se
s úspěchem využívá krystalové symetrie a je možné si vybrat jen několik
nejpravděpodobnějších prostorových grup, v kterých budeme hledat. Vybrané konformery je buď možné umisťovat jako fixní nebo jim nechat
nějaké stupně volnosti. Mřížkové parametry je možné v první přiblížení
nastavit tak, aby byli molekuly blízko sebe, ale nepřekrývali se. Hledání
krystalické formy s nejmenší energií je pak obdobné hledání vhodných konformerů. Ke klastrování se v tomto případě dá
využít i třeba podobnost práškových záznamů. Pro stabilitu dané krystalické
formy je podstatná Gibbsova volná energii G, která je daná součtem krystalové
energie E, součinu tlaku p s objemem V a záporně vzatý součin entropie S s teplotou T, viz rovnice (1). Člen p·V se dá snadno
spočítat, ale jeho vliv je při atmosférických tlacích malý. Výrazný problém je
vyjádření entropie. Tento člen se proto často zanedbává.
G = E + pV – ST (1)
Kromě predikování krystalových struktur ab-initio je možné využít výpočetní
metody také jako doplněk k neúplným či nekvalitním difrakčním datům. Např.
v práci [19] je popsáno predikování struktury s využitím neúplných
difrakčních dat měřených na vzorku za vysokých tlaků v diamantové cele.
V tomto případě tedy šlo o predikci s malým množstvím
experimentálních dat. Prakticky tyto data nemusí být difrakční a může jít i o
jiné experimentální výsledky. Pro difrakční komunitu může být energetický
výpočet nápomocný např. při lokalizaci vodíkových atomů, stabilizaci zpřesňování atomových pozic, či lokalizaci dvou podobně difraktujících prvků. Energetický výpočet by také mohl
urychlit konvergenci řešení struktur z práškových dat.
References
1. J. Maddox: Crystals from first
principles, Nature, 335 (1988), 6187
2. J. P. M. Lommerse. et
al, Acta Cryst. B56, 697-714, 10.1107/S0108768100004584
3. J. Kennedy;
R. Eberhart,: Particle swarm
optimization, Proceedings of ICNN'95 - International Conference
on Neural Networks,
(1995), 5263228
4. W. D. S. Motherwell. et al, Acta Cryst. B58, 647-661, 10.1107/S0108768102005669
5. G. Day. et al, Acta Cryst. B61,
511-527, 10.1107/S0108768105016563
6. G. Day, et al, Acta Cryst. B65,
107-125, 10.1107/S0108768109004066
7. D. Bardwell, et al, Acta Cryst. B67, 535-551, 10.1107/S0108768111042868
8. A.M. Reilly et al. Acta Cryst. B72,
439-459, 2016 10.1107/S2052520616007447
9. https://www.ccdc.cam.ac.uk/Community/initiatives/cspblindtests/csp-blind-test-7/
10.
Carlos M. Fonseca, Peter J. Fleming, Evolutionary Computation, 3 (1995), pp.1-16
11. https://cs.wikipedia.org/wiki/Optimalizace_hejnem_%C4%8D%C3%A1stic
12. M. Dorigo., C. Blum, Theoretical Computer Science, 344
(2005), pp. 243-278
13. Alessandro
Barducci, Massimiliano Bonomi, Michele Parrinello, Comput. Mol. Sci., 1 (2011), pp. 826–843
14. G.
G. Rondina and J. L. F. Da Silva, J. Chem. Inf. Model, 53
9 (2013), pp. 2282–2298
15. S. Goedecker, J. Chem. Phys., 120 (2004), 9911; https://doi.org/10.1063/1.1724816
19. N. Tsujimoto, D.. Adachi,
R. Akashi, S. Todo, and S. Tsuneyuki, Phys. Rev. Materials, 2 (2018), 053801