Application of paired refinement in fragment-screening project
M. Malý1,2, K. Diederichs3, J. Dohnálek2, P. Kolenko1,2
1Czech Technical University in Prague, Faculty
of Nuclear Sciences and Physical Engineering,
Břehová 7, 115 19 Prague, Czech Republic
2Institute of Biotechnology of the Czech
Academy of Sciences, Biocev,
Průmyslová 595, 252 50 Vestec,
Czech Republic
3University of Konstanz, Box M647, 78457 Konstanz, Germany
martin.maly@fjfi.cvut.cz
The paired refinement protocol [1] is considered as the optimal approach for the determination of high-resolution cutoff in macromolecular crystallography. Unlike the conservative criteria based only on the indicators of diffraction data quality, it enables a direct linking of the quality of data and structure model. Generally, the proper estimation of the resolution limit reduces the noise level in calculated electron density maps which subsequently leads to finer molecular structures. Especially, this has an impact on regions difficult to interpret.
During our recent work on PAIREF – automation of the protocol [2] – we analysed the data set from endothiapepsin from Cryphonectria parasitica in complex with fragment B53 [3], PDB entry 4Y4G. However, the mentioned ligand (molecule of fragment B53) is occupied only partially. Despite the importance of determination of exact molecule position, the signal is rather weak in this region.
Although the structure was previously solved at 1.44 Å resolution, we reprocessed the data in XDS [4] and performed paired refinement in PAIREF using REFMAC5 [5] up to 1.05 Å. Obtained results showed the data contain useful signal up to 1.20 Å. Moreover, the clear evidence of improvement of electron density quality in ligand region was observed while the optimal high-resolution cutoff was applied (Figure 1).
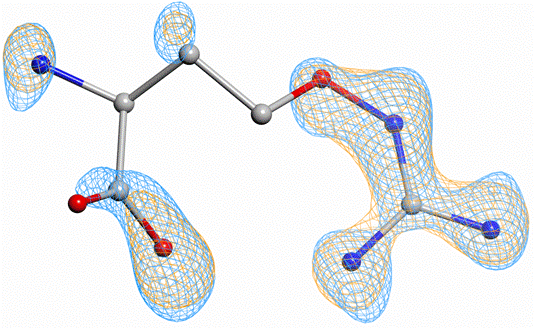
Figure 1. Comparison of electron-density omit maps of the partially occupied fragment B53 after refinement at 1.44 Å (orange) and 1.20 Å (blue) – contoured at 0.56 e·Å-3 level. Atomic coordinates originate from PDB entry 4Y4G [3]. The figure was created in CCP4mg [6].
1. P. A. Karplus & K. Diederichs, Science, 336, (2012), pp. 1030–1033.
2. M. Malý, K. Diederichs, J. Dohnálek, P. Kolenko, IUCrJ, 7, (2020), pp. 681–692.
3. F. Huschmann, J. Linnik, K. Röwer, M. Ühlein, X. Wang, A. Metz, J. Schiebel, A. Heine, G. Klebe, M. Weiss, U. Mueller, Acta Cryst. D, 72, (2016), pp. 346–355.
4. W. Kabsch, Acta Cryst. D, 66, (2010), 125.
5. G. N. Murshudov, A. A. Vagin,
E. J. Dodson, Acta Cryst. D, 53, (1997), pp. 240–255.
6. S. McNicholas, E. Potterton, K. S. Wilson, M. E. M. Noble, Acta Cryst. D, 67, (2011), pp. 386–394.
This publication was supported by the MEYS CR (projects CAAS – CZ.02.1.01/0.0/0.0/16_019/0000778 and BIOCEV – CZ.1.05/1.1.00/02.0109) from the ERDF fund and by the GA CTU in Prague (SGS19/189/OHK4/3T/14).