Možnosti počítačových simulací
J. Drahokoupil
Institute of physics, AS CR, Na Slovance 2, Praha 8, Czech Republic
draho@fzu.cz
Key words: structure solution, ab-initio calculation, molecular mechanics
Abstract
Personal experience with computational approach to crystal structure solution and refinement and also to crystal properties will be shortly summarized.
Úvod
V posledních letech se výkon a dostupnost počítačů neustále zlepšuje. Proto se predikování různých fyzikálních vlastností a struktury látek dostává do širšího podvědomí vědecké společnosti a buduje si postupně svou nepřehlédnutelnou roli ve vědeckém životě.
Pustil jsem se do počítačových simulací, aby mi „neujel vlak“, protože věřím, že podíl simulovaných výsledků vůči experimentálním se bude neustále zvyšovat. Navíc umožňují validaci a správnou interpretaci naměřených dat. Rád bych zde prezentoval několik příkladů využití, z kterými jsem měl možnost se setkat. Většina příkladů je počítána s využitím komerčního výpočetního balíku Material Studio [1] v rámci kterého zmíním moduly: CASTEP – kvantově mechanické výpočty pro periodické systémy v rámci DFT (Density functional theory), FORCITE a GULP – molekulová mechanika s možností editování či vytváření vlastních „forcefieldů“, REFLEX – zpracování difrakčních práškových dat, řešení i upřesňování struktury, SORPTION – sorpce malých molekul či atomů do porézních struktur. POLYMORF – predikce stabilních či metastabilních krystalických uspořádání dané molekuly. Program GULP je i samostatně a bezplatně dostupný [2]. Pro DFT výpočty jsem použil také program QUANTUM ESPRESSO [3] s grafickým rozhraním BURAI [4], který lze stáhnout zdarma.
Potenciální energie a geometrická optimalizace
Jednou ze základní úloh je výpočet potenciální energie dané struktury. Tuto úlohu lze pak následně rozvinout v hledání struktury s nejmenší energií. Ať už je použit klasický či kvantově mechanický přístup, je možné optimalizovat pozice atomů i velikost základní buňky. V případě hledání teoretické základní buňky nepřesahuje odchylka s porovnáním s experimentálními hodnotami několik málo procent. Na Obr. 1 jsou porovnány naměřené a teoreticky napočtené mřížkové parametry pro systém SrTi-MnO3. Struktury s částečnou či smíšenou okupancí jsou problematické. Řešením, je zavedení supercely, kde v dané pozici je jen den atom, ale průměrný počet daného atomu odpovídá stechiometrii.
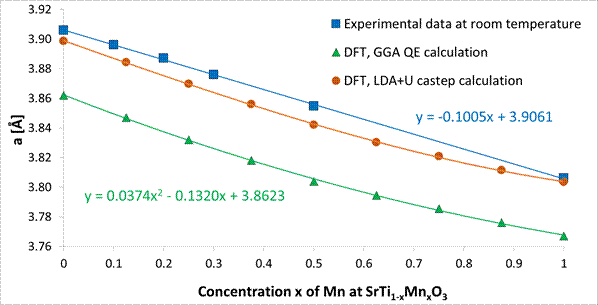
Obrázek 1. Mřížkové parametry v systému SrTi1-xMnxO3. Porovnání naměřených a ab-initio napočtených hodnot.
Upřesňování struktury z difrakčních dat s využitím energetického členu
V rámci modulu REFLEX je možné použít kombinované upřesňování struktury, při kterém je použit kombinovaný faktor shody
R = w RXRD + (1-w) Renergie, (1)
kde w je váha mezi difrakčním faktorem shody RXRD a energetickým faktorem shody Renergie. Jako difrakční faktor shody je použit klasický Rwp faktor. Energetický příspěvek je počítán pomocí modulu FORCITE a lze vyjádřit jako
Renergie = 0.1 (E – Emin)/ Etol, (2)
kde E je potenciální energie daného stavu, Emin je minimální nalezená potenciální energie a Etol je energetické toleranční okno.
Zavedení energetického členu vám drží molekulu pohromadě a je možné upřesňovat strukturu i pro relativně špatná difrakční data, či v případě kvalitních dat pokud jsme dále od globálního minima postupně zvyšovat váhový faktor ve prospěch difrakčního členu, jak se přibližujeme celkovému minimu. Výhodou oproti běžně používaným restrikcím je pružný charakter vazeb, který by měl umožnit snadnější pohyb atomů/molekuly směrem ke globálnímu minimu. Zavedení vlastních restrikcí a omezení je samozřejmě také možné.
SORPTION
V řadě aktivních forem zeolitů jsou některé atomy Si nahrazeny anionty Al-. Proto musejí být ve struktuře ještě nějaké kationty, které kompenzují tento náboj. Nalezení pozic těchto atomů ve struktuře pomocí difrakčních technik není jednoduché díky jejich částečné okupanci. Např. pro zeolit SSZ-16 jsou tyto atomy ve struktuře (66844-ICSD) umístěny do středu objemných kavit. Dle výpočtů tohoto modulu však vychází, že tyto Na+ kationty by měli ležet blízko Al- aniontů, tedy blíže hranicím jednotlivých kavit. Přesnější umístění těchto aniontů je zajímavé také z pohledu využití zeolitů jako molekulárních sít či absorbátorů. Je pak možné například předpovědět, kolik molekul metanu se vejde, při daném tlaku, do základní buňky daného zeolitu. V modulu SORPTION se používá přístupu klasické fyziky s použitím principů molekulové mechaniky. Velmi důležité je však správné přiřazení nábojů na jednotlivých atomech. Tento náboj je velmi často převzat z ab initio kvantově mechanických výpočtů.
Polymorfizmus a predikování struktur
Pro některé látky je téměř nemožné vypěstovat monokrystal, z kterého by se dala vyřešit struktura. Pro vyřešení struktury z prášku zase existují hranice, za které je velmi obtížné se dostat. Výpočetní postupy, které umožňují predikce struktury, proto nabízejí velmi zajímavou alternativní možnost. Modul POLYMORF opět vychází z klasické fyziky s vypočtenými náboji pomocí kvantového přístupu. Pokud máme flexibilní molekulu, která má vícero stabilních konformací, je nutné hledat vzájemné prostorové uspořádání těchto molekul pro každou z nich. Pro hledání stabilních konformací dané molekuly je možné využít modul CONFORMER, který umožňuje měnit nějaký stupeň volnosti, třeba torzní úhel z definovaným krokem a napočítá energie pro každou z těchto poloh, viz Obr.2.
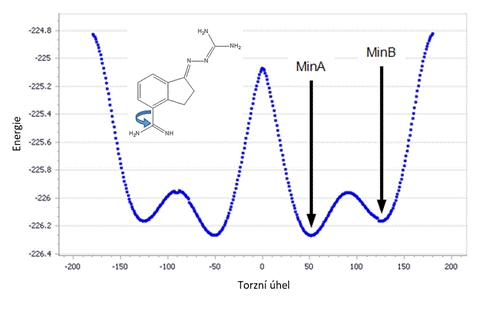
Obrázek 2. Energie v závislosti na torzním úhlu. Pro volnou molekulu jsou pozorovány dvě stabilní uspořádání MinA a MinB.
Po vybrání prostorové grupy/grup, v kterých chceme hledat řešení, se pustí Monte-Carlo simulace, která vygeneruje tisíce možných krystalových struktur. Výpočetně nejnáročnějším krokem je geometrická optimalizace a proto je možné tyto struktury nejprve roztřídit do skupin na základě vzájemné podobnosti a geometricky optimalizovat jen zástupce dané skupiny. Po geometrické optimalizaci následuje další klastrování, při kterém zjišťujeme, jestli některé potenciální struktury nespadly do toho samého energetického minima. Generování struktur na základě Monte-Carlo nezaručuje nalezení uspořádání s minimální energií, proto se doporučuje zopakovat celý proces několikrát. Možné polymorfy jsou ty, které mají nejmenší energii. Jsou to samozřejmě jen návrhy potenciálních prostorových uspořádání. Pokud máme k dispozici difrakční záznam, který nedosahuje takových kvalit, aby umožňoval stanovení struktury, je možné využít tento záznam k porovnání s difrakčními záznamy napočtenými z predikovaných struktur. Tato možnost je zajímavá např. v případě, že máte ve vzorku minoritní fází, která vám znemožňuje vyřešit strukturu klasickým způsobem. V případě porovnávání difrakčních záznamů hraje minoritní struktura vedlejší roli a dominantní struktura by měla být snadněji identifikovatelná.
Predikovatelné veličiny a vlastnosti
Co je na počítačových simulací z mého pohledu úžasné, že umožňují propojovat fyziku pevných látek. Je možné spočítat pásovou elektronovou strukturu, hustotu stavů, elektronovou hustotu, fononové disperzní křivky a hustotu stavů, elastické konstanty, bulk modulus, poissonovo číslo, optické a termodynamické vlastnosti, dielektrické a piezoelektrické konstanty a mnoho dalšího. Na Obr. 3 je do struktury SrTiO3 zakreslena izoplocha elektronové hustoty. Je patrné, že vazba Ti-O má oproti vazbě Sr-O kovalentní charakter.
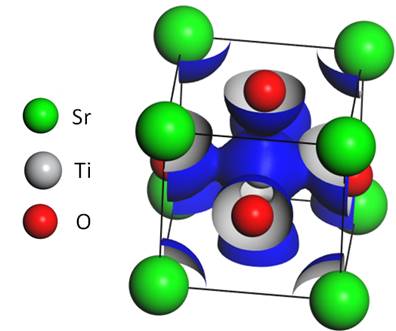
Obrázek 3. Struktura SrTiO3 a izoplocha elektronové hustoty.
Závěr
Počítačové simulace se stávají v současné době čím dál tím dostupnější, přesnější a otvírají nám krystalografům nové možnosti.
Literatura:
1. http://accelrys.com/products/collaborative-science/biovia-materials-studio/
2. http://gulp.curtin.edu.au/gulp/
3. https://www.quantum-espresso.org/
4. http://nisihara.wixsite.com/burai