Porovnávání solvátů organických látek programem CrystalCMP
J. Rohlíček
Oddělení strukturní analýzy, Fyzikální ústav AV ČR, v.v.i. Na Slovance
1999/2
182 21 Praha 8
rohlicek@fzu.cz
Podobnost mezi krystalovými strukturami může být definována na základě různých kritérií, např. podle symetrie, rozměrů základní buňky, podle složení nebo podle pozic atomů či molekul ve struktuře. Záleží vždy na tom, co je pro daný účel podstatné. Asi nejpoužívanějším rozdělením krystalových struktur na základě podobnosti je řazení do skupin podle tzv. strukturních typů [1], které se rozdělují na několik skupin. Tzv. izobodové (isopointal) jsou látky se stejným typem prostorové grupy a stejnými polohami atomů v buňce. Takto široce definovaná skupina látek může být dále rozdělena na látky izostrukturní (isoconfigurational), které mají podobné rozměry základní buňky a pozice atomů v buňce a mohou mít odlišné chemické složení. Dále existují látky krystalochemicky izotypní (crystal-chemically isotypic) a homeotypní (homeotypic). Vedle těchto zavedených pojmů lze definovat další a vytvářet tak nesčetné skupiny látek, které jsou si navzájem podobné podle různých kritérií.
V nedávné minulosti bylo publikováno několik různých metod, které určují podobnosti krystalových struktur na základě podobnosti rozmístění atomů v prostoru. Tyto metody můžeme rozdělit do dvou základních skupin – metody, které k porovnávání používají reprezentativní funkce, tzv. otisky prstu krystalové struktury [2]–[5]. V takových případech je jako otisk prstu vybrána vhodná funkce, např. párová distribuční funkce, která nese informaci o rozložení atomů v prostoru. Následně je podobnost mezi strukturami určena podle podobnosti jejich otisků prstu. Druhou skupinou jsou metody, které přímo porovnávají pozice atomů nebo celých fragmentů ve struktuře [6]–[10]. V těchto případech je nutné najít transformaci mezi porovnávanými strukturami, následně je překrýt a porovnat např. odchylky pozic atomů. Nalezení zmíněné transformace je nejtěžší úlohou a zároveň velikou výhodou těchto metod. V případě nejasností je možné porovnávané struktury přes sebe překrýt a visuálně překontrolovat.
Jedním z velkých problémů při porovnávání krystalových struktur mezi sebou je anisotropní expanze, např. kvůli různé teplotě během měření nebo kvůli přítomnosti atomů o různých atomárních poloměrech. Zajímavý přístup k řešení expanze krystalových struktur kvůli přítomnosti různých atomárních typů byl prezentován v Hund et al. (2006), kde jsou struktury nejprve seškálovány na stejnou hustotu a teprve poté je nalezena transformace a jsou porovnány pozice atomů mezi sebou.
V případě molekulárních krystalů je podobnost krystalových struktur studována pomocí podobnosti pakování celých molekul v prostoru. Mezi nejpoužívanější přístupy patří algoritmy implementované programem COMPACK [10] a xPac [11], [12]. Metoda programu COMPACK je implementována v programu Mercury [13] a je používána pod názvem Packing Similarity. Program vybere u každé porovnávané struktury reprezentativní molekulární klastr, kde je možné zvolit např. jen jeden typ molekuly (obvykle té největší ve struktuře), překryje je a následně provede porovnání na základě rozdílů pozic celých molekul v obou klastrech. Porovnání v případě programu xPac je prováděno tak, že se reprezentativním molekulárním klastru určí pakování dané molekuly v 1D, 2D a 3D směrech. Porovnání podobnosti reprezentuje podobnost pakování v 1D, 2D nebo ve 3D. Takže identické pakování je takové, které vykazuje podobnosti v 3D pakování, zatímco nejméně podobné struktury nevykazují podobnost pakování ani v jednom směru (1D).
Pracování s molekulárním klastrem místo základní buňky přináší několik skrytých, za to podstatných výhod. Především lze tímto způsobem porovnávat krystalové struktury, které krystalizují v různých prostorových grupách. Dokonce se tímto přístupem obchází omezení na podobnost tvaru a symetrie základních buněk. Lze tedy bez problému porovnávat např. triklinické struktury s kubickými.
Metoda programu CrystalCMP [14] je založena podobně jako metody programu xPac a COMPACK na porovnávání reprezentativního molekulárního klastru, do kterého je možné zahrnout jeden typ molekuly, obvykle té největší. Během porovnávání dojde k překrytí vygenerovaných molekulárních klastrů jednotlivých krystalových struktur a podobnost je spočítána jako odchylka středů překrývajících se molekul a úhlů natočení molekul od sebe. Výsledná podobnost (v tomto případě spíš rozdílnost) je dána vztahem
![]() , (1)
, (1)
kde Dc průměrná vzdálenost (v Å) středů překrývajících se molekul a Ad je průměrný úhel (ve stupních), který svýrají mezi sebou. Hodnota X je volena uživatelem a reprezentuje váhu mezi Dc a Ad (výchozí hodnota je X = 100).
Daleko větší váha je ve vzorci kladena na rozdíl natočení molekul v prostoru. Je to z toho důvodu, že stejné pakování není ani tak podmíněno stejnou pozicí molekul v prostoru, jako spíš jejich stejným natočením, viz. obr. 1. Právě zavedením druhého členu v Psab je umožněno porovnávání pakování molekul o relativně veliké expanzi spojené zejména s přítomností molekuly solventu o různé velikosti.
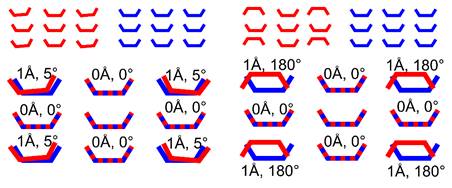
Obrázek 1. Grafické znázornění použité metody. Vlevo nahoře jsou dva téměř identické molekulární klastry (červený a modrý), kde je centrální molekula obklopena osmi sousedními molekulami. Po překrytí obou klastrů (vlevo dole) je vypočítána hodnota Psab = 0.5 + 100 × (2.5/180) = 1.9 (pro X = 100). Vpravo nahoře jsou také dva molekulární klastry, kde rozdíly pozic molekul jsou prakticky totožné, ale natočení molekul v prostoru je velmi odlišné – každá druhá molekula je převrácena o 180°. Po překrytí (vpravo dole) výpočet Psab = 0.5 + 100 × (90/180) = 50.5 (for X = 100) ukazuje na daleko vyšší rozdíl mezi porovnávanými klastry.
Výsledkem porovnání programem CrystalCMP je podobnostní matice a z ní vypočtený dendrogram, který seskupuje jednotlivé látky podle podobnosti v čitelné podobě. Program je napsán v jazyce C/C++, využívá knihovny OpenBabel [15] pro generování SMILES definic a pro grafické rozhraní používá wxWidgets a OpenGL. Program je volně šiřitelný a lze ho stáhnout z adresy http://sourceforge.net/projects/crystalcmp/, kde lze také nalézt jeho zdrojový kód.
Funkčnost programu je ukázána na studiu podobnosti pakování molekul ibrutinibu v jeho známých pevných formách. Doposud známé formy ibrutinibu [16], [17] jsou dva čisté polymorfy (v závorkách jsou uváděny kódy CSD databáze) – Forma A (BETXEG) a Forma C (BETWUV) a poté solváty anisolu (HAWLAV), chlorbenzenu (HAWLEZ), 1,3-dioxolanu (HAWLID), diacetonalkoholu (HAWLOJ), m-xylenu (HAWLUP), o-xylenu (HAWMAW), p-xylenu (HAWMEA), trifluorotoluenu (HAWMIE) a methanolu (RUYDEW01).
Z dendrogramu, viz Obr. 2, je patrné, že prakticky totožné pakování molekuly ibrutinibu lze nalézt v solvátech anisolu, m-xylenu a methanolu, viz. Obr. 3. Pakování ibrutinibu v těchto třech strukturách je pak ještě podobné pakování v dioxolan solvátu a ve formě C. Další podobnostní skupinu tvoří solváty chlorbenzenu, trifluorbenzenu, diacetonalkoholu a o-xylenu, viz. Obr. 4. Pakování molekuly ibrutinibu ve formě A a v p-xylen solvátu není podle Psab podobné s ostatními.
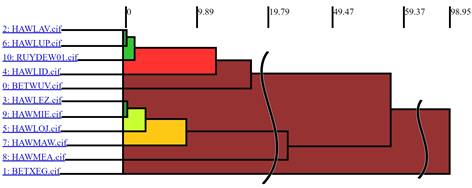
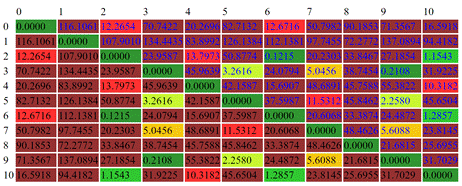
Obrázek 2: Výsledky porovnání pakování molekul ibrutinibu. V horní části je zobrazen dendrogram, kde na vodorovné ose je vynesena hodnota Psab funkce (menší hodnota znamená větší podobnost). Křivky znázorňují místa, kde byl dendrogram zkrácen. Svislá osa obsahuje CSD kódy jednotlivých forem ibrutinibu. V dolní části je pak zobrazena podobnostní matice s hodnotami Psab funkce, ze které byl dendrogram sestaven.
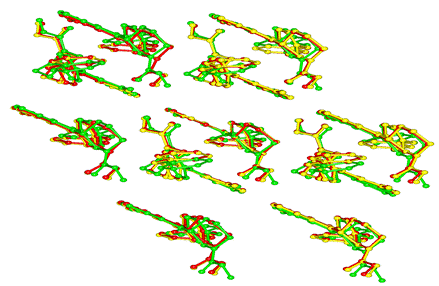
Obrázek 3: Porovnání pakování molekul ibrutinibu ve strukturách anisol solvátu (červená), m-xylen solvátu (žlutá) a methanol solvátu (zelená).
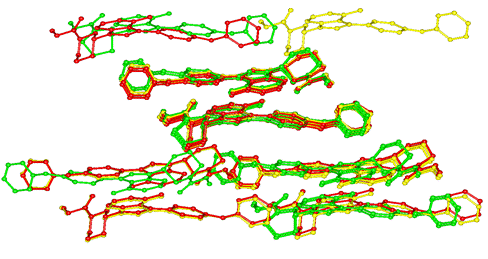
Obrázek 4: Porovnání pakování molekul ibrutinibu ve strukturách chlorbenzen solvátu (červená), trifluorbenzen solvátu (žlutá) a diacetonalkohol solvátu (zelená)
Program byl dále upraven tak, aby byl schopen provádět automatické porovnávání pakování molekul v zadaných krystalových strukturách. V současné době probíhá analýza celé CSD databáze. Pro každý záznam v CSD jsou nalezeny struktury obsahující stejné molekuly, kde molekula musí být největším fragmentem ve struktuře. Takto vytvořené seznamy jsou podrobovány analýze podobnosti pakování vybraných molekul a výsledky jsou ukládány ve formátu HTML. Vznikající stránka již dnes nese zajímavé informace o podobnosti pakování molekul v polymorfech, solvátech nebo i v látkách, které by nás ani nenapadlo mezi sebou porovnávat.
Rád bych touto cestou poděkoval grantu 17-23196S GAČR z jehož rozpočtu byl příspěvek na konferenci hrazen.