Validation of powder diffraction based structure determination results by DFT-D quantum mechanic calculations
M. Hušák1, J. Rohlíček2
1University of Chemistry and Technology, Prague, Technická 5, 166 28 Praha 6 – Dejvice
2Fyzikální ústav AV ČR, v. v. i., Na Slovance 1999/2, 182 21 Praha 8
husakm@vscht.cz
Introduction
Due to the loss of information related to measurement principle, results of crystal structure determination based on powder diffraction data are often not 100% reliable. Additional techniques like solid-state NMR, geometry check against common molecular parameters from databases or energy calculation should be used to verify the crystal structure and molecular geometry as well. The suggested methodology is described in details in article [1]. The mentioned article describes QM minimization of 225 molecular crystal structures determined by powder diffraction from witch 8.8% was found to be incorrectly solved. The aim of our work was to test whatever we can apply similar check to our results.
Methods
We did not have available VASP software nor the special force field method developed for dispersion correction implemented in GRACE software as used in the mentioned article [1]. Our calculations were done in CASTEP software as implemented in Material Studio [2] software package. We had used geometry optimization approach consisting of two steps as suggested in [1]: firstly, the cell was fixed during the geometry optimization, and secondly, cell parameters together with molecular geometry were optimized.
Details of Computation
For the system electron shield description, the GGA-PBE functional was used. The calculation setup was corresponding to "Fine" preset: 340 eV energy cut-off, convergence criteria - 10-5 eV/atom energy, 0.03 eV/Å maximal force, 0.001 Å maximal atom displacements. The Tkatchenko-Scheffler scheme was used for the dispersion correction.
The original powder based data were compared with the geometrically optimized data by the Cartesian displacement RMS (RMSCD) to be able to compare atomic positions in different unit cells. The Cartesian displacement for an atom in two crystal structures (1) and (2) is described by eq. (1).
![]() (1)
(1)
The ri are the fractional coordinates of atoms in crystal structure i and Gi is the transformation matrix from fractional to Cartesian coordinates for structure i. The RMSCD calculation was implemented in CrystalCMP software [3].
Results
The obtained results are summarized in Tab. 1.
|
CSD code |
RMSCD for non H atoms (Å), cell fixed |
RMSCD for all atoms (Å), cell fixed |
RMSCD for non H atoms (Å), cell refined |
RMSCD for all atoms (Å), cell refined |
|
BOVDUM |
0.2652 |
0.3762 |
0.2931 |
0.4024 |
|
SORLOC |
0.1093 |
0.2026 |
0.1073 |
0.213 |
|
TISCAB |
0.0932 |
0.3145 |
0.0937 |
0.3146 |
|
WAFLIA |
0.0947 |
0.2542 |
0.0947 |
0.2542 |
|
XOGLOW |
0.1346 |
0.2044 |
0.1367 |
0.2091 |
|
XOGLUC |
0.1069 |
0.1668 |
0.1107 |
0.1747 |
Table 1. RMSCD values for 6 tested structure
Discussion
According to the [1], structures with RMSCD > 0.25 Å for non H-atoms should be considered as suspicious and checked. The BOVDUM structure falls in such category. The issue can be related to disorder - for the QM calculation only one part of the disorder, which was observed in the crystal structure, was used. However, it sounds like the explanation is different. This crystal structure (capacitabine) was later re-determined by other group from single crystal [4] data and it is presented in CSD with code BOVDUM01. The structure redetermination had observed an incorrect placement of H-atom in our original powder study, see Fig. 1.
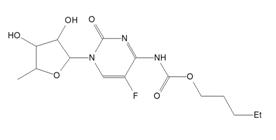
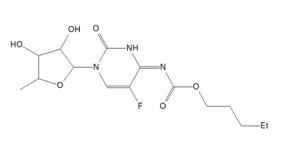
Figure 1. Comparison of incorrect (left) and correct (right) BOVDUM (capecitabine) formula in crystalline state
The TISCAB structure shows low value of RMSCD for non-H atoms, but much higher value for all atoms including hydrogen atoms (RMSCD >0.3!). The QM structure suggests a different H atom position and bond order as described on Fig. 2. . The original article [5] suggests the formula based on liquid NMR data only. In solid state, the formula can be different. A search in CSD did not find any similar crystal structure with the same order of hydrogen atoms as in [5], but it had found about 30 structures with hydrogen atoms in positions suggested by the QM.
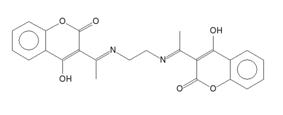
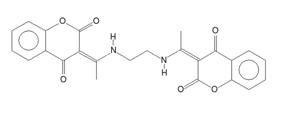
Figure 2. Comparison of incorrect (left) and correct (right) TISCAB formula in crystalline state
Conclusions
In two from six our powder studies, the DFT-D calculation had found troubles mainly with positioning of hydrogen atoms. It is even worse than the 19/215 issues as found in [1] but the total number of structure checked is too low to make a reasonable statistics and conclusions. It is probably clear anyway that any molecular crystal structure solved from powder should be checked by DFT-D geometry calculation before publications to detect possible issues.
1. J. Streek, M. A. Neumann, Acta .Cryst., B70, (2014), 1020.
2. http://accelrys.com/
3. https://sourceforge.net/projects/crystalcmp/
4. M. Malinska, P. Krzeczyński, E. Czerniec-Michalik, K. Trzcińska, P. Cmoch, A. Kutner, K. Woźniak, J. Pharm. Sci.,103,(2014), 587.
5. J. Rohlicek, I. Kateta, T.B. Ayad, R.B. Hassen, J. Mol. Struct.,. 1051, (2013), 280.
We would like to thank the Grant Agency of the Czech Republic for financial support (Grant No. 106/14/03636S).