Dynamic Theory of Protein Crystallization as a Tool for a Control of Protein Crystal Architecture
Jindřich Hašek
Institute of Biotechnology CAS, BIOCEV, Průmyslová 595, 252 50 Vestec, Czech Republic
hasekjh@seznam.cz
Crystallography is very precise and handy method for determination of “small molecular structures”. In structure determination of biological macromolecules and their complexes, the processing is much more complex and the precision is much worse. The crystallization process is considered as hardly predictable and thus crystallization trials are based on an expensive random screening of many hundreds of crystallization conditions as a rule. Nevertheless, the success rate of crystallization of the native proteins is still quite low. A deeper understanding the crystallization process seems to be prerequisite for radical improvement of the present status.
Many problems with protein crystallization are caused by obligatory high water content in crystals (50 % on average). Protein molecules remain in crystals highly solvated and are in dynamic equilibrium with solution also in crystalline state. The stability of molecules in crystal is thus ensured only by intermolecular interactions between adhesive patches of neighbor macromolecules forming a rigid 3D scaffold in the crystal. Most of solvent filling 30-80 % of crystal volume remains dynamically disordered (“fluent”) and invisible in maps of electron density. Thus, the rigidity and stability of the 3D macromolecular scaffold of crystal is of basic importance, i.e. the design of the protein crystal architecture is very important.
Dynamic theory of protein crystallization. Promising approach seems to change the traditional paradigm believed by now that the naked protein molecules are depositing to form the crystalline state. New dynamic theory of protein crystallization supposes that protein molecules form temporary low affinity adducts with some additives present in successful crystallization screens forming thus temporary protein coat that can be varied simply by composition of crystallization solution. The adhesion properties of the coated proteins (temporary adducts) may be completely different then the adhesion properties of the naked proteins. Then, the coated proteins deposit completely different way than the untouched protein molecules. Energy stabilizing the protein coat should be low in comparison with the adhesion energy of the protein-protein adhesion mode dominating the crystal growth. Therefore, protein molecules lose their envelopes during deposition of next protein molecules into the crystal scaffold, i.e. the temporary adducts disintegrate. The molecules forming the protein coats are gradually being squeezed out from growing crystals and diffuse by concentration gradients into solution. As a result, one usually obtains a crystal of the pure native protein without crystallization additives.
Existence of these temporary adducts in crystallization solutions is experimentally well documented, in spite of the fact that the direct observation of small molecules movement during crystallization process on molecular level is very problematic. The dynamic theory of protein crystallization may be difficult for imagination, but having good knowledge of the protein-protein adhesion, and of the protein-additive adhesion, one gets a straight way for rational control of protein crystallization and for rational design of the desired protein crystal architecture.
Large surface of protein molecules offers usually many adhesive patches and thus we can design different architectures of the protein scaffold (polymorphism). Each crystal form has its own set of compatible adhesion modes, the sets of incompatible adhesion modes and its own optimal solvent content in the crystal. When different protein-protein adhesion modes are mixed together in the same protein aggregate, one gets almost amorphous precipitant.
Principle of a single dominating adhesion mode. As the definition of crystal requires periodic repetition of a single structure motif, we can formulate the following postulate for successful protein crystallization. The principle of a single dominating adhesion mode plays a key role in controlling the diffraction quality of crystal. It says that well diffracting crystals grow only when a single adhesion mode dominates during crystallization, and when all adhesion modes incompatible with the dominating adhesion mode (DAM) are suppressed. Any molecules deposited in incompatible adhesion mode depreciate significantly diffraction quality, and can lead up to virtually non-diffracting solid phase.
The best protein crystal architecture can be tuned by rational composition of crystallization solution using the protein surface active molecules (PSAM) and the protein surface shielding agents (PSSA] [3]. They provide us with active (used in the crystal scaffold) and passive (shielding) elements (leaving during crystallization) used in a design of the best protein crystal architecture for our protein.
Using PSAM to mutate adhesion properties of protein molecules is much easier than other ways of changing adhesion modes of protein, e.g. chemical modification of the protein surface, lysine methylation, fragmentation of protein (or protein complex) into smaller crystallizable parts, or complexation with auxiliary high affinity molecules possessing higher crystallization propensity. Namely, using PSAM, one gets the structure of the natural protein as the final result and not the structure of some artificial construct.
Role of PEG in crystallization. The dynamical theory of protein crystallization and knowledge of protein-protein adhesion modes explains why the poly(ethylene glycol)-type polymers (PEGs) are the most successful precipitants [1,2]. The reason is that the PEG-type polymers have, in addition to their precipitating effect, also a large scale of PSAM and PSSA properties. Analysis of these properties is described in our former papers [1,2]. Reader can also get his own experience using the tools offered by the database of protein-polymer interactions. He can scan over hundreds selected protein structures and observe readily various protein-PSAM adhesion modes. The Table with categorized protein-PSAM interactions can serve as a guide to evaluate applicability of the useful adhesion modes (in press).
The analysis of already solved protein structures deposited in the PDB gives the necessary knowledge of adhesion properties of proteins required for planned strengthening the dominant and weakening of other non-compatible adhesion modes in crystallization solution. These studies including a review of protein crystal architecture (in press) can be simplified by the “Database of protein-polymer interactions”.
Database of protein-polymers interactions (DPPI) [2] contains about 4000 experimentally observed PEG-protein interfaces. It consists of a set of protein structures crystallizing with PEG-like polymers and useful scripts allowing easy visualization of PEG activities on protein surfaces. Seeing the PEG fragments interfering with protein-protein interfaces, with different types of salts, blocking competitive crystal contacts helps us to understand the dynamic processes during crystal growth and allows rationalization of crystallization screens on molecular basis. The database of protein-polymer interactions is available from hasekjh@seznam.cz.
An example of PEG used as active construction elements (PSAM) is shown in the Figure bellow. Here, the PEG chains are non-standardly built into the protein skeleton and are important for architecture of this special crystal form. They stick firmly the neighbor protein molecules. However, they can mutually slide without excessive loss of energy. This type of connection allows the complementary adhesion modes fully use their adhesive potential without evoking not desired stress in the crystal.
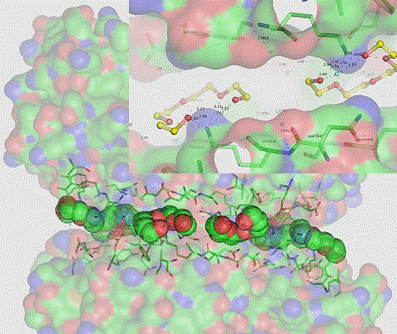 |
Figure 1. An example of an active construction element (PSAM) useful in building the crystalline scaffold. The PEG induced adhesion mode is here a leading motif. The insert shows a typical crown ether conformations of four polyether (PEG) chains induced by interaction of five ether oxygens to positive charge of lysine exposed on the surface of neighbor protein molecules. The lysines induce the crown-ether like conformation of polyether with high number of CH2 groups exposed into solvent. As a result, the two neighbor protein molecules are joined by strong hydrophobic interactions. We call this construction element as flexible glue because hydrophobic interactions allow a mutual slide of the two protein molecules allowing thus a high degree of flexibility of the crystal scaffold. PDB code 3NBT.
Conclusion. Dynamic theory of crystallization, the principle of the domination adhesion mode, knowledge of the adhesion properties of protein surface active molecules to proteins, and also knowledge of standard protein-protein adhesion modes gives us an efficient tool for:
· crystallizing most of proteins including those supposed non-crystallizable by now,
· growing the crystal polymorphs offering the best accuracy of structure determination,
· observation of proteins in different hydration degrees and different molecular environments,
· analyzing experimentally different protein-protein adhesion modes. Some of these adhesion modes are undoubtedly of the basic importance for processes in living cells.
1. Hašek J., Z.Kristallogr. 28, (2011), 475-480.
2. Hašek J. et al, Z.Kristallogr. 23, (2006), 613-619.
3. Hašek J. et al, J.Synchr.Radiation 18, (2011), 50-52.
The project is supported by BIOCEV CZ.1.05/1.1.00/02.0109 from the ERDF, CSF project 15-15181S of the CSF, grant LG14009 of MSMT, BioStruct-X (EC FP7 project 283570) and Instruct of ESFRI.