Isopolyoxometalate functionalisation facilitated by transition metal complexes
E. Rakovský
Comenius University, Faculty of Naturel Sciences,
Department of Inorganic Chemistry, Mlynská dolina CH2–212, Ilkovičova 6, 842 15
Bratislava, Slovak Republic
rakovsky@fns.uniba.sk
Polyoxometalates has a long record of interesting structures [1] and potentially exploitable properties in the area of nanotechnology, catalysis, magnetic properties [2]. Incorporation of paramagnetic, photochemically or photophysically active ions make them valuable components of materials. Antitumoral, antiviral and antibiotic properties of several POMs and specific POM–protein interactions were also studied [3].
In contrast with the relative high stability of hetero-POMs, the chemistry of isopolyoxometalates is more challenging. Vast majority of iso-POMs exists only in narrow pH and c(M) regions, often with the POM cores allowing to obtain several protonation modes. In solutions, there are complex protolytic (and eventually, redox) equilibria with many species present in reaction mixtures. The crystallisation of often marginally soluble products can be also tricky.
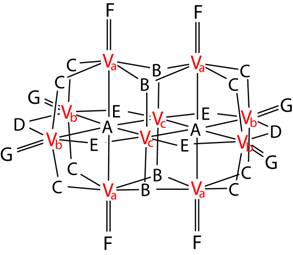
Figure 1. The scheme of the decavanadate anion with idealized D2h point group geometry. A–G denote crystallographically non-equivalent O atoms and Va–Vc denote crystallographically non-equivalent V atoms (based on [4]).
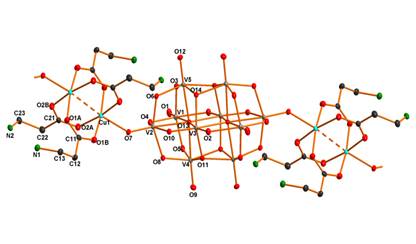
We studied several decavanadate-based systems (Fig. 1) based on transition metal ions (CuII [5, 8, 9], MnII [6, 7], ZnII [7], CoII) and proposed ligands (b-alanine bAla [5, 7], 2-(2-hydroxyethyl)pyridine hep [6, 9], glycine gly [8], 2-(aminomethyl)pyridine amp [9]). Initially, we obtained one of the first compounds containing decavanadate as a bridging ligand (and first 1D coordination polymer), (NH4)2[Cu2(bAla)4(V10O28)]·10H2O (1, Fig. 2). Coordination of the paddlewheel Cu(II) complex cation to the centrosymmetrically arranged G sites of the decavanadate core is assisted by strong hydrogen bonds. The attempts to prepare similar decavanadate complexes with bAla ligand lead to the preparation of the noncoordinated decavanadates (NH4)2[M(H2O)5(NH3CH2CH2COO)]2V10O28·nH2O (M = ZnII, n = 4; M = MnII, n = 2). Although both containing [M(H2O)5(NH3CH2CH2COO)]2+ cation, these cations are structurally different.
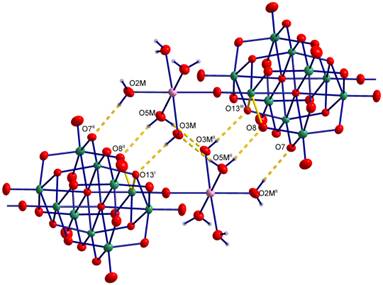
In attempt to prepare hep based complexes, in the system with MnII, hep is acting only as a counterion hepH+ to the pendant complex with the [Mn(H2O)5]2+ complex unit coordinated to the centrosymmetrically arranged F sites of the bridging decavanadate anion in the (hepH)2[{Mn(H2O)5}2V10O28]·4H2O (2, Fig. 3). Complex anions forms the supramolecular chains via the hydrogen bonding network consisting of aqua ligands.
|
|
|
Figure 2. The 1D chain motif in the structure of 1. |
Figure 3. Supramolecular anionic chains in the structure of 2. |
|
|
|
Figure 4. The anionic unit of 3. |
Figure 5. The structure of the neutral complex 4. |
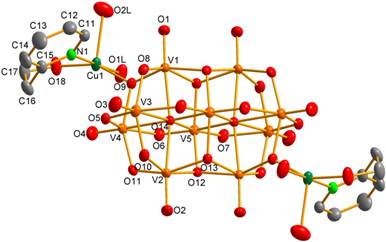
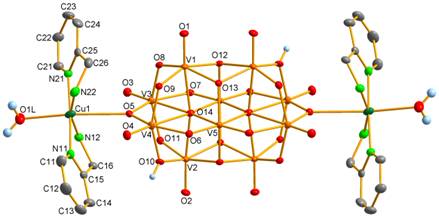
From the reaction system containing CuII and hep, the complex with bridging decavanadate ligand coordinated at the centrosymmetricaly arranged C sites by the two [Cu(H2O)2(O,N-hep)]2+ units, (hepH+)2[{Cu(H2O)2(O,N-hep)}2V10O28]·6H2O (3, Fig. 4), was prepared.
To illustrate coordination possibilities of the decavanadate core, from the same system with the amp used instead of hep, the [{Cu(amp)2(H2O)}2H2V10O28] ·4H2O (4, Fig. 5) was prepared. If in the 3 we had shortest (V–)O–M bond ever found (1.9563(16) Å), in the 4 it is the longest one (2.665(2) Å). It is due to coordination of the cationic unit to the centrosymmetrically arranged D sites of the anion, which are hard to access.
From the system with Cu(II) and glycine, (NH4)2[Cu2(H2O)4(gly)2 (gly–)2]H2V10O28·6H2O was prepared. The compound contains dinuclear copper complex with bridging water ligands along the dihydrogendecavanadate anion. The interesting feature of the cation is that it contains both O-coordinated glycine a N,O-coordinated glycinato ligands.
1. A. Dolbecq, E. Dumas, C. R. Mayer, and P. Mialane, Chem. Rev., 110, (2010), 6009–6048.
2. U. Kortz, A. Müller, J. van Slageren, J. Schnack, N. S. Dalal, and M. Dressel, Coord. Chem. Rev., 253, (2009), 2315–2327.
3. B. Hasenknopf, K. Micoine, E. Lacôte, S. Thorimbert, M. Malacria, and R. Thouvenot, Eur. J. Inorg. Chem., (2008), 5001–5013.
4. J. L. F. da Silva, M. F. M. da Piedade, and M. T. Duarte, Inorg. Chim. Acta, 356, (2003), 222–242.
5. L. Klištincová, E. Rakovský, and P. Schwendt, Inorg. Chem. Commun., 11, (2008), 1140–1142.
6. L. Klištincová, E. Rakovský, and P. Schwendt, Acta Crystallogr. C., 65, (2009), m97–9.
7. L. Klištincová, E. Rakovský, and P. Schwendt, Transit. Met. Chem., 35, (2010), 229–236.
8. L. Klištincová, E. Rakovský, P. Schwendt, G. Plesch, and R. Gyepes, Inorg. Chem. Commun., 13, (2010), 1275–1277.
9. L. Bartošová, Z. Padělková, E. Rakovský, and P. Schwendt, Polyhedron, 31, (2012), 565–569.
This work has been supported by the Ministry of Education of Slovak Republic (Grant VEGA 1/0336/13).