Určování amorfního podílu v částečně
krystalických materiálech
M. Čerňanský
Fyzikální ústav AV ČR, v. v. i., Na Slovance 2, 182 21 Praha 8, Česká republika
cernan@fzu.cz
Stanovení krystalického a amorfního podílu je společným problémem u všech materiálů, ve kterých mohou koexistovat amorfní a krystalické fáze. Obsah amorfních fází v neúplně krystalických látkách je důležitou charakteristikou těchto materiálů, protože řada fyzikálních a technologických vlastností značně závisí právě na amorfním podílu. Difrakční analýza je jedinou přímou metodou k určení podílu krystalických a amorfních fází. Podstatou této metody je rozdělení difrakčního záznamu na části odpovídající krystalickým fázím a na části odpovídající amorfní fázi. Přitom však působí velmi nepříznivě ta skutečnost, že difrakční záznam obsahuje řadu rušivých informací, jakou je záznam difrakce spojitého spektra primárního svazku, rozptylu primárního záření na vzduchu, okrajích clon vymezujících primární svazek, nekoherentního rozptylu atd.
K eliminaci zmíněných parazitních vlivů je zpravidla nutné provést další měření bez měřeného vzorku za jinak naprosto stejných podmínek. Dodržení stejných experimentálních podmínek je nutné také při kalibraci měření. Numerické určení procenta krystalické nebo amorfní fáze v měřeném vzorku totiž vyžaduje srovnání s nějakým standardem. Zde se často užívá další úplně krystalický vzorek. Kromě dalšího měření za naprosto stejných podmínek však úplně krystalický standard může vést i k jiným problémům. Především u některých materiálů může být velmi obtížné, ne-li nemožné, připravit úplně krystalický vzorek, který by obsahoval fáze se stejnou strukturou, jaké jsou v částečně zkrystalizovaném vzorku. Během krystalizace měřeného vzorku totiž mohou vznikat metastabilní fáze, které se nakonec v úplně krystalickém standardu vůbec nevyskytují.
Jedním z řešení je použití úplně amorfního standardu [1]. Zde však je důležité to, aby se chemické složení amorfní fáze během krystalizace neměnilo. V opačném případě by se intenzita difrakce od amorfní fáze měnila nejen proto, že se mění její zastoupení ve vzorku, ale také díky změně jejího chemického složení. To je zvlášť nežádoucí v případech, kdy se v amorfní fáze mění obsah těžkých prvků, které mají velkou rozptylovou schopnost [2].
Dalším možným řešením je použití některé z metod, vycházejících ze strukturně nezávislých naměřených a vypočtených charakteristik difrakce [3]. Je nutné zdůraznit, že všechny způsoby stanovení amorfního podílu vyžadují velmi přesná měření a mimořádně pečlivou eliminaci parazitních vlivů. Použití klasické fotografické detekce difraktovaného záření u těchto způsobu není vhodné. Všeobecně se doporučuje použít monochromatické záření, pokud je to možné taky vakuovou komůrku a výpočtem provést korekci na nekoherentní rozptyl [4].
Splnění výše zmíněných požadavků se zatím realizovalo především sestavením vhodných experimentálních zařízení. Jednalo se o Goppelovu komůrku s fotografickou registrací difraktovaného záření [5], sestávající z větší měřící komůrky, uvnitř které byla menší kalibrační komůrka. Analogii Goppelovy komůrky s elektronickou registrací záření [5] navrhl Alexandr se spolupracovníky. Další uspořádání s difraktomtrem a monitorem navrhli Krimm a Stein [6]. I v současnosti, díky zobrazovacím foliím, může být velmi užitečná komůrka navržena Kochanovskou a Sochůrkem [7], umožňující zhotovení řady snímků za přesně stejných expozičních a vyvolávacích podmínek.
Z teorie kvantitativní fázové analýzy je známé, že intenzita difrakčních maxim je v prvním přiblížení primo úměrná koncentraci příslušné krystalické fáze ve směsi a totéž platí pro intenzitu koherentního rozptylu amorfní fázou [2]. Vzhledem k tomu, že koeficienty této úměrnosti jsou pro různé fáze (a pro různé reflexe) jiné, neudává poměr intenzit krystalické a amorfní fáze přímo numerickou hodnotu amorfního podílu. Často se však definuje stupeň krystalinity R a index krystalinity Xc jako podíl intenzit difrakčních maxim od krystalické (Ic) a amorfní (Ia) fáze [8]
R = Ia /Ic , Xc = Ic /( Ic + K Ia ) = 1 / (1 + KR), (1)
kde R je poměr intenzit amorfní a krystalické faze a K je konstanta. Pro relativní porovnání může být položena rovna 1. Do vztahů (1) se obvykle dosazují integrální intensity, pokud by intensity maximální nebyly přesnější.
K převodu veličiny Xc na absolutní procento krystalické xc, resp. amorfní xa fáze ve vzorku je nejlepší použit standard úplně krystalický, nebo úplně amorfní. V principu je možné použít vzorek se známým obsahem amorfní nebo krystalické fáze za podmínky, že struktury amorfní a krystalické fáze v měřeném vzorku i ve standardu jsou stejné. Existují také metody určující amorfní podíl současně u dvou [9] nebo více vzorků [10] umožňující získat absolutní procento amorfní nebo krystalické fáze bez standardů.
V principu existují dva základní způsoby na určení amorfního podílu xa [2]. V tom prvním se určí koncentrace všech krystalických fází, většinou metodou vnitřního standardu. Amorfním podílem je pak rozdíl součtu koncentrací krystalických fází od jedničky. Při druhém způsobu určení obsahu amorfní fáze se určuje veličina ca přímo. Tento způsob je založen na skutečnosti, že intenzita koherentního rozptylu od amorfní faze je přímo úměrná jejímu obsahu ve vzorku. Křivka amorfního rozptylu obsahuje jedno nebo několik rozmytých maxim, obvykle v oblasti nižších úhlů rozptylu. Měření intensity v tomto případě vyžaduje náročnou experimentální techniku, protože na rozptyl amorfní fáze se superponuje rozptyl spojitého spektra vzorkem, rozptyl vzduchu, nekoherentní rozptyl a difuzní rozptyl od tepelného pohybu atomů a od jejich statických vysunutí z pravidelných poloh. Je žádoucí použít krystalový monochromátor.
Na přímé určení amorfního podílu jsou v podstatě dvě metody – s pomocí standardů nebo bezstandardová. S pomocí standardu se amorfní podíl určí podle vztahu [11]
ca = 1 – [(Ia0 - Ia)/(Ia0 - Ib)], (2)
kde Ia0 je intenzita rozptylu úplně amorfním standardem při určitém pevném úhlu 2Θ, Ia je intenzita rozptylu sledovaným, měřeným vzorkem pod stejným úhlem a Ib je intenzita rozptylu směsi krystalických fází, která má chemické složení stejné jako amorfní standard. Úhel 2Θ se vybírá tak, aby rozptyl amorfní fáze byl dost výrazný a současně aby krystalické fáze neměly v daném místě nějakou reflexi. Měření vzorku neobsahujícího amorfní fázi je nutná k započtení pozadí způsobeného výše zmíněnými příčinami.
Bezstandardová metoda na přímé určení amorfního podílu - podobně jako celá obecná teorie určování amorfního podílu - je založena na zákoně zachování intenzity rozptylu [12], [13], který tvrdí že intenzita koherentního rozptylu zprůměrována přes celý reciproký prostor, nezávisí na struktuře objektu. Pokud tedy je intenzita dopadajícího rentgenového záření konstantní, je integrální intenzita záření koherentně rozptýleného stejným množstvím látky stejná, bez ohledu na její vnitřní strukturu, zejména bez ohledu na to, jaký je poměr množství krystalické a amorfní faze.
K experimentálnímu určení amorfního podílu existuje několik metod. Metoda Goppela a Arlmana [5] je založena na vztahu
xa = [(1/t’)(Ia’/IR’)]/[(1/t)(Ia/IR)], (3)
kde - Ia, resp. Ia’ je intenzita amorfního maxima úplně, resp. částečně amorfního vzorku,
- IR, resp. IR’ je intenzita reflexe standardu odpovídající intenzitě Ia, resp. Ia’,
- t, resp. t’ je tloušťka úplně, resp. částečně amorfního vzorku.
Metoda určení indexu krystalinity [8]
vychází přímo z jeho definičního vztahu (1), kdy se určí podíl intenzity
difrakčního maxima krystalické faze a součtu intenzit
difrakčních maxim amorfní a krystalické faze. Předpokladá se, že lze snadno rozlišit intenzity
difraktované od amorfní a od krystalické části vzorku. K převodu na absolutní procento lze využít např. měření
specifických objemů amorfní fáze Va, krystalické faze Vc
a měřeného vzorku Vx [8]
xc = [(Va - Vx) / (Va – Vc)] x 100 % , (4)
Poslední vztah tak umožňuje kalibraci mezi relativní veličinou Xc a absolutní veličinou xc, t. j. určit K v (1).
Metodu měření diferencí intenzity [5], [8], [13] lze použít v případě širšího měřeného intervalu, ve kterém se vyskytuje několik výrazných difrakčních maxim. Rozlišení na difrakci od amorfní faze a krystalické faze je v tomto případě zpravidla těžce proveditelné. Změří se proto dva referenční vzorky – jeden s velmi vysokou krystalinitou (krystalický standard - c) a druhý s velmi nízkou krystalinitou (amorfní standard - a) z téže látky jakou je měřený vzorek - u. Ve stejných bodech téhož intervalu měření se určí diference mezi normalizovanými hodnotami intenzit : (Iu - Ia) a (Ic - Ia). “Integrální” index krystalinity je pak definován vztahem
Xc = [ ∑ (Iu - Ia)] / [ ∑ (Ic - Ia) ] , (5)
kde sumace probíhají podle řady vybraných hodnot difrakčních úhlů 2Θ ve zvoleném intervalu. K převodu na absolutní procento krystalické, resp. amorfní fáze je nutné další měření, podobně jako u předchozí metody. Metoda měření diferencí intensity má kromě již zmíněné integrální verze ještě korelační modifikaci. Závislost hodnot y = (Iu - Ia) na x = (Ic - Ia) se aproximuje přímkou, jejíž směrnice Xc je “korelační” index krystalinity.
Metoda Hermanse a Weidingera [5], [9] je opět vhodná v případě kdy lze rozlišit výrazné difrakční maximum krystalické fáze od difrakce fáze amorfní, ale v tomto případě je výsledkem absolutní hodnota, resp. absolutní procento krystalické a amorfní faze. Základním předpokladem je, že se struktura fází vznikajících při krystalizaci nemění. Pak je možné užít ke kalibraci další vzorek, který nemusí být ani úplně krystalický ani úplně amorfní, dokonce nemusí být u něj známý podíl krystalických a amorfní fáze. Označíme-li totiž x jako krystalický podíl v prvním měřeném vzorku a y krystalický podíl v druhém měřeném vzorku, je zřejmě (1 - x), resp. (1 - y) podíl amorfní fáze v prvním resp. druhém vzorku. Číslo A = x/y pak může znamenat podíl integrálních intenzit dané difrakce krystalů u obou měřených vzorků a číslo B = (1 - x)/(1 - y) se určí spíše jako podíl výšek prvního difrakčního maxima od amorfní fáze u obou měřených vzorků, protože hodnoty integrálních intenzit u amorfní fáze by mohly být zatíženy většími chybami. Uvedené definice čísel A, B lze považovat za soustavu dvou rovnic pro dvě neznámé x, y, jejichž řešením vychází
x = (A – AB)/(A - B), y = (1 – B)/(A – B) (6)
Z toho lze v principu určit absolutní procento krystalických a amorfních fází obou vzorků. Vyžaduje to však opět
přesná měření za naprosto stejných podmínek u obou vzorků.
Regresní metoda [5], [10] spočívá v proložení přímky přes body o souřadnicích x = Ia , y = Ic odpovídajících jednotlivým vzorkům. Tato přímka vytíná na ose Ic hodnotu I100c odpovídající úplně krystalickému vzorku a podobně na ose Ia hodnotu I100a odpovídající úplně amorfnímu vzorku. Označíme-li K = I100c / I100a, vychází pro absolutní hodnotu krystalického podílu [5]
xc = 1 / [1 + (K/R)] , (7)
kde R je stupeň krystalinity. Předností této metody je, že nepotřebuje žádný standard. Je jen nutné mít několik vzorků s různým, i když neznámým, stupněm krystalizace.
Rulandova metoda [5], [8], [14] respektuje skutečnost, že difuzní rozptyl od teplotního pohybu atomů a od jejich statických vysunutí z pravidelných poloh, t. j. od nedokonalostí mřížky v krystalické části vzorku, nelze jednoduše odečíst od naměřených intenzit. Ruland [14] proto navrhl, aby se při výpočtu celkové intenzity od krystalické části vzorku použily strukturní faktory FD, které by zohledňovaly odchylky atomů z jejich pravidelných poloh. Formálně to znamená, že v příslušných rovnicích se namísto čtverců atomových rozptylových faktorů f 2 objeví F2D , což lze popsat zavedením funkce neuspořádánosti D. Pro nedokonalosti krystalové mřížky prvního druhu, u kterých se zachovává pořádek na dlouhou vzdálenost, navrhl Ruland [15] tvar D = exp (- ks2), což dobře popisuje neuspořádanost od tepelného pohybu atomů a jejich statických vysunutí z pravidelných poloh. Pro nedokonalosti mřížky druhého druhu, u kterých se pořádek na dlouhou vzdálenost ztrácí, jako je to např. u parakrystalů, se doporučuje [15]
D = [2 exp (-as2)] / [1 + exp (- as2)]. Pro krystalický podíl pak namísto
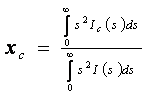 vychází vztah
vychází vztah 
kde středování čtverců atomových rozptylových faktorů je provedeno přes různé druhy atomů.
Metoda založena na strukturně nezávislých charakteristikách rozptylu vychází z průběhu J(Θ) intensity difraktované amorfní částí vzorku, který je nutné určit z difrakčního záznamu. Následně se spočte
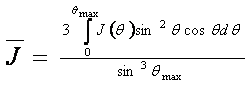 a
a 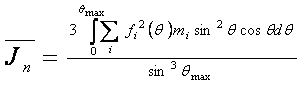
kde Θmax je maximální hodnota úhlu Θ dosažena v experimentu. Levá veličina souvisí s amorfní fází a pravá, spočtena pomocí zákona o zachování intensity, je celková intenzita nezávislého rozptylu všech atomů vzorku bez interference. Podíl levé a pravé veličiny se rovná amorfnímu podílu.
[1] L. Červinka & J. Dusil, J. Non-Crystal. Solids, 21 (1976) 125-136.
[2] L.S. Zevin & L.L. Zavjalova: Količestvennyj rentgenografičeskij fazovyj analiz, Moskva 1974. Nedra. s. 42-45.
[3] A.S. Kagan & V.M. Snovidov, Zav. Lab., 31 (1965) 573-576.
[4] L.I. Mirkin: Spravočnik po rentgenostrukturnomu analizu polikristallov, Moskva 1961. Fizmatgiz. s. 27-28.
[5] L.E. Alexander: X-Ray Diffraction Methods in Polymer Science, New York 1969. Wiley. pp. 137-197.
[6] S. Krimm & R.S. Stein, Rev. Sci. Instrum., 22 (1951) 920-925.
[7] A. Kochanovská & A. Sochůrek, Papír a celuloza, 9 (1954) 84-87.
[8] J.E. Spruiell & E.S. Clark: in Methods of Experimental Physics: Polymers., Vol. 16, Part B, ed. R.A. Fava. New
York 1980. Academic Press. pp. 114-127.
[9] P.H. Hermans & A. Weidinger, J. Appl. Phys., 19 (1948) 491-506.
[10] G. Challa, P.H. Hermans & A. Weidinger, Makromol. Chem., 56 (1962) 169-178.
[11] S.M. Ohlberg, & D.V. Strickler, J. Am. Ceram. Soc., 45 (1962) 170-171.
[12] B.K. Vajnštejn: Difrakcija rentgenovych lučej na cepnych molekulach, Moskva 1963. Izdatělstvo AN-SSSR.
[13] M. Kakudo & N. Kasai: X-Ray Diffraction by Polymers, Tokyo – Amsterdam 1972. Kodansha – Elsevier.
[14] W. Ruland, Acta Cryst., 14 (1961) 1180-1185.
[15] W. Ruland, Polymer, 5 (1964) 89-102.