VYUŽITÍ VÝPOČETNÍCH METOD PŘI STUDIU STRUKTURY
J. Drahokoupil
Institue of Physics AS CR, Na Slovance 2, Prague 8, 182 21, Czech Republic.
draho@fzu.cz
Počítačové simulace v pevných látkách dosahují v
posledních letech velký rozmach. Jejich aplikace umožňuje dobrou pomoc při
řešení struktury pevných látek v případech, kdy nejsou k dispozici dostatečně
dobrá difrakční data. Podíváme se na dva přístupy: i) časově náročnější výpočty
s využitím ab-initio DFT (Density Function Theory), ii) rychlejší výpočty s
využitím molekulové mechaniky.
DFT
Základním kamenem pro teorii DFT je teorém
Hohenbergera a Kohna [1], později zobecněný Levym [2], který tvrdí, že všechny
vlastnosti v základním stavu jsou funkcionálem nábojové hustoty ρ. Konkrétně energie může být
napsána jako:
Et[ρ] = T[ρ] + U[ρ] + Exc[ρ] (1)
kde T[ρ] je kinetická energie systému
neinteragujících částic s hustotou ρ,
U[ρ]
je klasická elektrostatická energie Coulombových interakcí a Exc[ρ] zahrnuje všechny mnohočásticové příspěvky k totální energii
(výměnou a korelační energii). Pro vyjádření posledního členu, použijeme tzv.
Local Density Aproximacion (LDA), která je založena na známé Exc pro jednoelektronový plyn
[3]. LDA
předpokládá, že nábojová hustota se na atomové úrovni jen pozvolna mění.
Pomocí DFT lze vypočítat řadu vlastností pevných
látek, pro nás nejzajímavější jsou parametry základní buňky a souřadnice atomů.
Na rozdíl od difrakce není možné uvažovat částečné zastoupení dané pozice
atomem. Atom buď na dané pozici je nebo není. Pro popsání substitučních pozic
či vakancí je nutné příslušně zvětšit základní buňku. Další nevýhodou je že se
počítá tzv. základní stav tedy pozice a mřížka odpovídající 0° K. Přes tyto
nevýhody dosahuje přesnost výpočtů v porovnání s experimentálními hodnotami
shody několika málo procent.
Molekulová
dynamika
Na rozdíl od DFT, která vychází z kvantové teorie,
vycházejí modely molekulové mechaniky z klasické fyziky. Vzájemné působení
atomů je popsáno pomocí silových konstant. Celková potenciální energie systému
je vyjádřena jako součet vazebných a nevazebných interakcí, jejich funkce a
parametry jsou shrnuty v takzvaném silovém poli (forcefield). Parametry
silových polí lze získat buď výpočtem pomocí kvantové mechaniky či porovnáním
výsledků z experimentálních hodnot (difrakce, NMR, rotační a vibrační
spektroskopie, elastické konstanty,…). Tento jednodušší formalizmus umožňuje
řešit podstatně větší problémy, už se nemusíme omezovat na jednotky či desítky
atomů, ale jdou řešit problematiky obsahující stovky či tisíce atomů. Vhodný
náhled do dané problematiky lze nalézt zde [4].
Unikátní spojení molekulové mechaniky a difrakce je
zavedení faktoru shody Rcomb
kombinující difrakční (např. Rwp)
a energetický faktor RE.
Rcomb = (1-w) Rwp + w RE (2)
Optimální hodnotu váhového faktoru w je pak možné volit na základě tzv.
Pareto optimalizace, kde se napočtou a porovnají hodnoty parametrů shody. Na osu
x se obvykle vynáší Rwp faktor, který ukazuje
shodu s difrakčními daty a na osu y
hodota E – Emin, viz obr, 1.
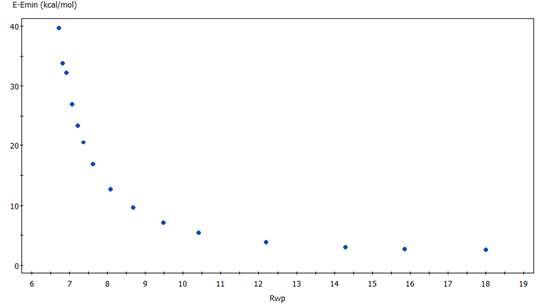
Obrázek 1. Pareto optimalizace umožňuje vybrat optimální
váhový faktor mezi energetickým a difrakčním příspěvkem do společného faktoru
shody Rcomb. Jednotlivé body v
tomto konkrétním grafu odpovídají z leva doprava postupnému nárůstu váhového
faktoru w.
V
přednášce budou předvedeny konkrétní výpočty v programovém balíku Material
Studio [5] s využitím modulů Castep (DFT), Forcite
(molekulová mechanika) a Reflex (prášková difrakce). Poslední zde uvedený modul
umožňuje navíc kombinovat právě prášková rentgenová difrakční data spolu s
výpočty pomocí molekulové mechaniky.
References
1. P. Hohenberg, W. Kohn: "Inhomogeneous electron gas", Phys. Rev. B, 136, 864-871 (1964).
2. M. Levy: "Universal variational functionals of electron densities, first-order density matrices, and natural spin-orbitals and solution of the v-representability problem", Proc. Natl. Acad. Sci. U. S. A., 76, 6062-6065 (1979).
3. D.M. Ceperley,
B.J. Alder: "Ground state of the
electron gas by a stochastic method", Phys Rev. Lett., 45, 566-569 (1980).
4. P. Kovář, M. Veteška:
"Výpočetní postupy v molekulární
mechanice" Materials Structure,
vol. 19, no. 2, 75-79 (2012).
5. http://accelrys.com/products/materials-studio/
Acknowledgements.
Tato práce byla
podpořena projektem MPO FR-TI 2/165.