Výpočetní postupy v molekulární dynamice
M. Pospíšil, M. Veteška
Univerzita Karlova v Praze, Ke Karlovu 3, 12116
Praha 2
pospisil@karlov.mff.cuni.cz
Keywords: molecular dynamics, statistical ensemble, thermostat,
pressure control
Abstract
The general procedures used in
molecular dynamics are described. Conditions of calculations under constant
temperature and constant pressure in different statistical ensembles for
various thermodynamics quantities are presented. Methods of molecular dynamics
for the global minimum search are mentioned.
Molekulární dynamika
Molekulární
dynamika [1-3] je založena na změnách poloh atomů v závislosti na silách,
které na ně působí – jedná se o výpočet klasické pohybové rovnice. Síly se vypočítají
z potenciální energie určené na základě parametrů silového pole. Molekulární
dynamika je vhodná ke studiu časového vývoje systémů za nenulové teploty.
Hlavní aplikace molekulární dynamiky jsou:
- Hledání konformací
– během simulace se mění uspořádání systému, a tak lze zkoumat různé části
stavového prostoru, které jsou pro model dostupné.
- Generování
statistických souborů – použitím technik na kontrolování teploty a tlaku lze
generovat statistické soubory, z nichž lze vypočítat různé energetické,
termodynamické, strukturní a dynamické vlastnosti.
- Studium pohybu
molekul – např. uzpůsobení se molekul do specifických tvarů, chemické reakce
nebo odvození difúzních koeficientů.
Integrační algoritmus
Newtonova pohybová
rovnice pro atom i se souřadnicemi ri je
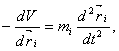 (1)
(1)
kde V je potenciální
energie. Tato rovnice je deterministická, tj. pro zadané úvodní souřadnice a
rychlosti se dají pozice atomů určit v kterémkoli čase. Tyto určené souřadnice
a rychlosti pro celou počítanou dynamiku se nazývají trajektorie. Rovnice se
řeší metodou konečných diferencí, kdy ze známých souřadnic a rychlostí v čase t jsou spočteny
souřadnice a rychlosti v čase t + Dt. Pro molekulární
dynamiku je vhodný algoritmus, který je rychlý (potřebuje na jeden krok ideálně
jen jedno vyhodnocení energie, protože to je pro velké modely časově velmi
náročné), potřebuje málo paměti, umožňuje použít dlouhý časový krok Dt a dobře zachovává energii (zejména aby bylo možno
generovat korektní statistické soubory).
Taková kriteria
nejlépe splňuje Verlet-leapfrog algoritmus. Odvodí se pomocí rozepsání pohybové
rovnice (1) s vhodným vyjádřením argumentů:
![]()
![]() (2)
(2)
![]() (3)
(3)
Tyto rovnice se
přepíší jako numerické symetrické derivace
![]() (4)
(4)
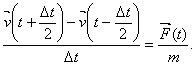 (5)
(5)
Vlastní algoritmus
začíná nastavením v čase t poloh r(t) a v čase t − Dt/2 rychlostí v(t − Dt/2) Poté se provádí kroky:
1. Výpočet síly v
čase t
![]() (6)
(6)
2. Přepočtení
rychlostí v čase t + Dt/2
![]() (7)
(7)
3. Přepočtení poloh
v čase t + Dt
![]() (8)
(8)
4. Posunutí času t = t + Dt a pokračování prvním krokem.
Výhodou tohoto algoritmu je právě jeden výpočet síly na jeden časový krok,
malá spotřeba paměti a relativně dlouhé časové kroky. Nevýhoda je, že polohy a
rychlosti jsou vypočteny v polovičním kroku od sebe. Tento problém nemá
Verletův rychlostní algoritmus, ten ovšem potřebuje navíc paměť pro uložení
vypočtených sil z předešlého kroku. V případě potřeby přesnější trajektorie se
větší chyby Verletovy metody dají kompenzovat průměrováním přes sadu
trajektorií. Jiné, přesnější algoritmy, např.Rungeho–Kuttaova metoda se obvykle
nepoužívají, neboť vyžadují v každém kroku několikanásobný výpočet sil. Podrobnosti o uvedených algoritmech lze nalézt např.
v [4 – 6].
Časový krok se volí
co nejdelší, ovšem příliš dlouhé kroky zapříčiňují nestabilitu a nepřesnost
výpočtu. V případě molekulárních simulací je třeba zvážit také omezení modelu,
které je dáno zejména pohybem atomů s nejvyššími frekvencemi (vodík), a zvolit
takový krok, aby byl splněn předpoklad, že během jednoho kroku jsou rychlosti a
zrychlení konstantní.
Statistické soubory
Výpočet pohybových
rovnic prochází plochu konstantní energie systému. Nicméně v přirozeném případě
je systém většinou vystaven vnějšímu tlaku nebo vyměňuje teplo s okolím a
celková energie se nezachovává. Existují metody, jak rozšířit molekulární
dynamiku, aby se dala teplota a tlak kontrolovat. V závislosti na tom, které
stavové veličiny se zachovávají konstantní, se dají generovat různé statistické
soubory.
NVE soubor
Jedná se o tzv.
mikrokanonický soubor, je adiabatický (tzn. k výměně tepla nedochází),
konstantní je celková energie a objem. Získá se standardním řešením Newtonových
rovnic. K fluktuacím energie dochází jen díky zaokrouhlovacím chybám, tedy
tento soubor je vhodný, když je třeba prozkoumat povrch konstantní energie nebo
když je potřeba se vyhnout fluktuacím vznikajícím kontrolou teploty a tlaku.
Teplota fluktuuje okolo průměrné hodnoty v závislosti na tom, jak se přeměňuje
energie mezi kinetickou a potenciální energií.
NVT soubor
Jde o tzv.
kanonický soubor, je izotermický, tzn. dochází k výměně tepla s termostatem,
konstantní je termodynamická teplota a objem. Používá se buď pro systémy, u
nichž tlak není potřebný faktor, aby se jeho ignorováním zmenšily fluktuace
trajektorie, nebo pro systémy bez periodických okrajových podmínek, protože u
těchto systémů tlak nelze definovat.
NPT soubor
Konstantní je
teplota i tlak. Použitelný je jen pro periodické systémy, neboť kontrola tlaku
se provádí změnami velikosti mřížové buňky. Je vhodný např. k nastavení
rovnováhy s požadovanou teplotou a tlakem před následnou simulací v NVE
souboru.
Kontrola teploty
Termodynamická
teplota je makroskopická stavová veličina, která má smysl pouze pro systém v
rovnováze. Teplota má k mikroskopickému popisu systému vztah přes kinetickou
energii, tj. přes rychlosti atomů. Maxwellovo–Boltzmannovo rozdělení popisuje v
rovnovážném systému o teplotě T pravděpodobnost f(v), že atom o hmotnosti m má velikost
rychlosti v:
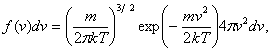 (9)
(9)
kde k je Boltzmannova
konstanta. Velikosti kartézských složek rychlosti vx mají Gaussovo
rozdělení pravděpodobnosti g(vx)
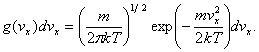 (10)
(10)
Termodynamická
teplota se měří z průměrné kinetické energie systému, s kterou souvisí přes
ekvipartiční teorém, který říká, že každému stupni volnosti přináleží průměrná
energie kT/2. Tzv. kinetická teplota Tkin a okamžitá kinetická
energie K tak
mají vztah
![]() (11)
(11)
kde Nf je počet stupňů
volnosti a N je
počet atomů s rychlostmi vi. Termodynamická teplota T je průměrem
kinetické teploty Tkin, tedy platí
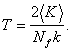 (12)
(12)
Počet stupňů
volnosti Nf bývá v neperiodických systémech 3N − 6, protože každý atom má 3 složky rychlosti, od čehož je
6 stupňů odečteno, neboť rotace a translace těžiště jsou ignorovány. V
periodických systémech jich je obvykle 3N - 3, protože se ignorují jen
translace těžiště.
Ačkoliv na začátku
simulace se atomům přiřadí rychlosti podle Maxwellova–Boltzmannova rozdělení
při dané teplotě, během simulace rozdělení nezůstává stejné. To se děje zejména
tehdy, když simulace nezačíná ze struktury, která je zcela minimalizovaná.
Rovněž se během simulace mění teplota, když se minimalizovaná struktura mění ve
strukturu v teplotní rovnováze a kinetická energie přechází v potenciální
energii.
Konstantní teplota
se udržuje algoritmy, které musí upravovat rychlosti atomů tak, aby dohromady
dávaly teplotu korektně, tedy podle Maxwellova–Boltzmannova rozdělení. K tomu
je třeba navíc zaručit vytváření korektního statistického souboru, tj. že
pravděpodobnost výskytu jisté konfigurace se chová podle statistických zákonů.
Např. pro soubor NVT to znamená, že pravděpodobnost výskytu konfigurace s
energií E je
úměrná tzv. Boltzmannovu faktoru exp(−E/kT).
Přímé přeškálování
rychlostí
Jde o drastický
způsob změny rychlostí atomů tak, aby bylo přesně dosaženo žádané teploty.
Ačkoli je to velmi efektivní způsob, během simulace se obyčejně nepoužívá,
protože neprodukuje korektní statistický soubor, potlačuje přirozené fluktuace
systému. Používá se k rychlé změně teploty během snahy dostat systém do
rovnovážného stavu s požadovanou teplotou.
Berendsenův
termostat
Tato metoda lze
použít pro rovnovážný systém a dává již dobré přiblížení kanonickému souboru.
Každá rychlost je znásobena faktorem
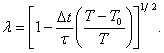 (13)
(13)
kde Dt je délka kroku, τ je charakteristický relaxační čas, T0 je požadovaná teplota
a T je okamžitá
kinetická teplota [7].
Noseho–Hooverův
termostat
Tato metoda je
složitější a je třeba použít Hamiltonián systému. Výsledkem je přesný kanonický
soubor. V metodě se pro reprezentaci interakce s termostatem přidává k modelu
fiktivní stupeň volnosti s vhodnou hmotností a korektním potenciálem. Výpočet
pohybových rovnic s konstantní energií (tj. v NVE souboru) pro rozšířený systém
pak vede k NVT souboru pro reálný model [8-11].
Andersenova
metoda
Tato metoda je
založena na tom, jakým způsobem je ovlivňována teplota v reálném systému. Jedna
verze Andersenovy metody změní v každém kroku rychlost jednoho atomu podle
Boltzmannova rozdělení. Jiná verze náhodně mění s předem danou kolizní
frekvencí rychlosti všech atomů.
Kontrola tlaku
Tlak je
termodynamická stavová veličina. Počítá se využitím viriálového teorému [12].
Okamžitý tlak P při
kinetické teplotě T, objemu systému V , v němž je N částic, je
![]() (14)
(14)
W je vnitřní viriál
![]() (15)
(15)
kde ri jsou pozice atomů a
fi jsou síly na ně
působící. Termodynamický tlak je průměrem okamžitého tlaku.
Tlak může být
definován jen v případě, kdy je systém v nějaké buňce o daném objemu. V případě
molekulových simulací je to mřížová buňka, proto lze tlak spočíst jen pro
periodické modely. Stejně jako v případě teploty musí metody na kontrolu tlaku
vytvářet korektní statistické soubory. Metody jsou podobné jako u teploty.
Berendsenova
metoda
V této metodě je
tlak upravován změnou souřadnic částic a velikosti buňky, a to tak, že v každém
kroku jsou jak všechny tři souřadnice každého atomu, tak vektory mříže znásobeny
stejným faktorem
![]() (16)
(16)
v němž Dt je krok, P je okamžitý tlak, P0 je žádaný tlak a γ a τ jsou volitelné parametry
stlačitelnost a relaxační čas. U této metody se projevují výrazné fluktuace a
není ji vhodné obecně používat [13].
Andersenova
metoda
V této metodě je
objem brán jako proměnná systému. K Lagrangiánu systému jsou přidány dva členy:
člen reprezentující kinetickou energii mříže, který má jako parametr volitelně
definovanou hmotnost, a člen reprezentující elastickou energii, jež je součin PV tlaku P a objemu buňky V. Z tohoto
Lagrangiánu se odvozují pohybové rovnice pro atomy a mřížové vektory [14].
Pokud je třeba
simulovat za neizotropního tlaku, používá se Parrinellova–Rahmanova metoda [15],
která je obdobná Andersenově metodě.
Obecný postup
dynamických výpočtů
Dynamické simulace
probíhají ve dvou fázích: dosažení rovnovážného stavu s požadovanými podmínkami
a vlastní simulace (sbírání dat, produkční fáze). Každá z nich představuje
samostatnou dynamickou simulaci.
Za rovnovážný stav
se považuje konfigurace systému, která nejvíc odpovídá požadované teplotě a
tlaku. Jeden ze způsobů jak poznat, zdali bylo rovnováhy dosaženo, je měřit v
závislosti na čase termodynamické veličiny, jako jsou energie, teplota, tlak. V
rovnováze tyto veličiny fluktuují okolo svých průměrných hodnot, které jsou v
čase konstantní. Ovšem zcela dostatečný test to není, neboť konfigurace se může
náhle změnit i po dlouhé době. Jistější způsob ověření rovnováhy je začít
výpočet z různých počátečních konformací s různými rychlostmi. Pokud
dokonvergují k podobným konformacím a vlastnostem, bylo pravděpodobně rovnováhy
dosaženo.
Na začátku fáze
hledání rovnováhy se atomům přiřadí rychlosti podle Maxwellova–Boltzmannova
rozdělení a během hledání rovnovážného stavu je možno teplotu udržovat i přímým
přeškálováváním rychlostí. Pokud je systém velký, může trvat poměrně dlouho
najít rovnovážný stav, protože je třeba prohledat rozsáhlý stavový prostor. Na
dobu hledání rovnováhy má rovněž negativní vliv, pokud jsou velké energetické
bariéry mezi lokálními minimy a globálním minimem.
Následná simulace
po dosažení rovnováhy již slouží k zaznamenávání údajů o systému. Teplotu
v této fázi nelze kontrolovat přímým přeškálováváním rychlostí. K dosažení
rovnováhy je možno použít jiný statistický soubor než pro následnou produkční
fázi a je samozřejmě možné z jednoho rovnovážného modelu pokračovat ve
více simulacích za rozdílných podmínek.
Nalezení globálního minima
Lze předpokládat,
že model s nejmenší celkovou energií bude pravděpodobně také ten
nejrealističtější. Proto cílem molekulárního modelování je nalézt globální
energetické minimum. To se provádí změnami všech stupňů volnosti, jimiž jsou
pozice atomů, rozměry mříže atd. Zmíněné klasické minimalizační algoritmy (viz.
P. Kovář: Výpočetní metody v molekulární mechanice) většinou dokáží najít
jen lokální minima, která jsou blízko výchozí konformace, neboť tyto algoritmy
ignorují konformace, jejichž energie je vyšší. Jedině procházením celého
povrchu energie, který je dán zmíněnými stupni volnosti, lze nalézt globální
minimum. Pro nalezení globálního
minima je třeba mít metody pro generování velkého množství výchozích modelů s
rozdílnými konformacemi. Tyto metody lze rozdělit podle přístupu do třech
skupin:
1. deterministické systematické procházení celého
povrchu energie,
2. stochastické metody (např. Monte Carlo,
genetické algoritmy),
3. molekulární dynamika.
Pochopitelně nejspolehlivějšími metodami je
procházení celého povrchu energie, tj. prostoru všech možných stavů např. krystalu.
Toto je možné pouze pro malé modely, neboť pro velké struktury významně narůstá
výpočetní čas, a je třeba použít další dva přístupy.
Nutno ještě dodat, že hledat globální minimum má
smysl zejména u malých molekul (dosažení globálního minima lze poznat tak, že
minimalizační algoritmy k němu dokonvergují z vícero rozdílných výchozích
konformací). U větších struktur často existuje několik různých konformací,
které jsou v lokálním minimu a mají obdobnou energii, a tudíž se mohou všechny
vyskytovat. Metody na hledání globálního minima pak místo toho slouží k
nalezení všech takto možných stavů.
Deterministické procházení stavového
prostoru
Tyto metody spočívají ve vygenerování mnoha
hrubých výchozích konfigurací, tzv. iniciálních modelů, z nichž ty nejvhodnější
jsou následně minimalizovány. Aby bylo opravdu nalezeno globální minimum,
iniciální modely musí pokrývat celý prostor možných stavů. U různých konformací
molekuly se příliš neliší délky a úhly vazeb, ale torzní úhly. Proto konkrétní algoritmus
výběru iniciálních modelů obyčejně spočívá v systematických změnách všech
torzních úhlů molekuly.
Stochastické metody
Tyto metody nedokáží obecně prohledat celý stavový
prostor, ale většinou se jedná o vytváření nových iniciálních modelů z již dříve
minimalizovaných modelů s vložením náhodných malých změn. Tyto iniciální modely
jsou pak zase standardně minimalizovány.
Nejčastějšími metodami jsou torzní Monte Carlo,
kartézské stochastické metody nebo na genetických algoritmech založené metody,
které využívají celé sady výchozích minimalizovaných modelů, jež jsou postupně
vylepšovány jejich vzájemným ovlivňováním.
Typy molekulární dynamiky
Molekulární dynamika lze použít i za účelem
vytváření nových konformací. Požadavek rovnovážnosti není třeba a na vhodné
procházení prostoru stavů se používají jiné, odpovídající techniky dynamiky. Molekulární
dynamika využívá kinetické energie atomů k překonání potenciálových bariér. To
jí umožňuje dosáhnout stavů molekul, které jsou pro minimalizaci nedostupné.
Délka dynamiky bývá nejvýše několik nanosekund, což je příliš málo na významnou
změnu konformace. Proto se molekulární dynamika hodí k efektivnímu prozkoumání
lokálního prostoru stavů, ale nelze ji v tomto případě užít k hledání
globálního minima. Toto omezení se snaží překonat některé speciální techniky
dynamiky, používající případně vyšší teploty (lze použít i klasickou dynamiku
za vyšší teploty, která umožňuje překonat velké energetické bariéry). Lze
využít např. následující typy dynamik:
„Quenched”
dynamika (zhášená)
Tato technika nezvyšuje možnosti dynamiky nalézt
globální minimum, ale prochází dosažitelný prostor stavů užitečněji. Je to
kombinace klasické dynamiky a minimalizace. Pravidelně, po jistém počtu kroků
dynamiky je struktura minimalizována. Poté dynamika pokračuje od stavu před
minimalizací. Minimalizované stavy se zaznamenávají odděleně od stavů
dosažených během dynamiky. Tuto metodu lze kombinovat s jinými technikami
dynamiky.
Dynamika
„simulated annealing“ (žíhací)
V této technice se teplota ve zvolených krocích
zvyšuje z původní k cílové teplotě a poté zase zpět. Tento cyklus se
případně několikrát opakuje. Zvýšení teploty umožní překonat i vysoké
energetické bariéry.
Při kombinaci s Quenched dynamikou se minimalizuje
stav s nejmenší energií během posledního cyklu.
Impulsní
dynamika
Impulsní dynamika umožňuje před spuštěním výpočtu
přiřadit výchozí rychlosti o určitém směru vybraným atomům. Takto lze rovněž překonat
energetické bariéry.
References
1. P. Comba, T. W. Hambley, Molecular Modeling of
Inorganic Compounds. VCH, Weinheim, 1995.
2. M. Veteška, Molekulární simulace ve strukturní
analýze interkalátů. MFF UK Praha 2006.
3. Cerius2 User Guide,
Forcefield Based Simulations. Molecular Simulations Inc., San Diego, 2000.
4. W. H. Press, B. P. Flannery, S. A. Teukolsky, W. T.
Vetterling, In Numerical Recipes, The Art of Scientific Computing. Cambridge
University Press, Cambridge, 1986.
5. L. Verlet, Phys. Rev., 159, (1967), 98 –
103.
6. I. Nezbeda, J. Kolafa, M. Kotrla, Úvod do
počítačových simulací metody Monte Carlo a molekulární dynamiky, Karolinum,
Praha 1998.
7. H. J. C. Berendsen, J. P. M. Postma, W. F. van
Gunsteren, A. DiNola, J. R. Haak, J. Chem. Phys., 81, (1984), 3684-3690.
8. S. Nosé, Molec. Phys., 52, (1984), 255-268.
9. S. Nosé, J. Chem. Phys., 81, (1984),
511-519.
10. S. Nosé, Prog. Theoret. Phys. Supplement, 103,
(1991), 1-46.
11. W. Hoover, Phys. Rev. A, 31, (1985),
1695-1697.
12. M. P. Allen, D. J. Tildesley, Computer Simulation
of Liquid, Claredon Press, Oxford Science Publications, 1987.
13. D. Brown, J. H. R. Clark, Molecular Physics, 51,
(1984), 1243-1252.
14. H. C. Andersen, J. Chem. Phys., 72, (1980),
2384-2393.
15. M. Parrinello, A. Rahman, J. Appl. Phys., 52,
(1981), 7182-7190.