Semi-automatické hledání
ligandů v proteinových strukturách
F. Pavelčík1,
J. Václavík2
1Ústav chemických léčiv, 2Ústav
přírodních léčiv
Farmaceutická fakulta,
Veterinární a farmaceutická univerzita v Brně, Palackého 1-3, 61242 Brno
pavelcikf@vfu.cz
Keywords: Protein-ligand complex, rotation
function, translation function, ligand docking
Úvod
Automatizované hledání
ligandů v protein-ligandových komplexech je žádané v některých moderních
metodách designu léčiv jako je např. FBDD (fragment-based drug design). FBDD je relativně nová metoda [1] objevování
léčiv. Místo hledání celého léčiva se hledají nejprve jednotlivé malé fragmenty,
které mají doplňkový povrch a vysokou interakční energii k aktivnímu místu
cílového proteinu. Výběr cílových proteinů byl umožněn současným dramatickým
rozvojem metod molekulární biologie, genetiky a příbuzných disciplin.
Principiální průlom v designu léčiv je vyžití fyzikálních metod screeningu jako
je NMR nebo rtg. krystalografie místo farmakologického screeningu (HTS).
Princip FBDD metody je velmi jednoduchý a je zobrazen na obr. 1. Malé fragmenty
se následně spojí chemickými metodami do větší molekuly, tak aby prostorové
rozložení funkčních skupin bylo stejné jako ve struktuře protein-ligandových
komplexů. Klasické farmakologické nebo biochemické metody obyčejně vůbec nejsou
schopné detekovat požadovanou biologickou aktivitu těchto malých fragmentů.
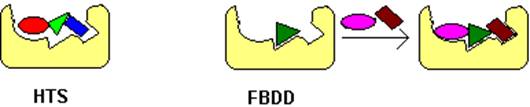
Obr. 1. Princip fragmentové metody designu léčiv.
V rtg. metodách se
používá koktejl 4-10 malých molekul z knihovny obsahující řadově 100 sloučenin.
Pokud by byla cílová molekula složena ze tří fragmentů je taková knihovna
ekvivalentní knihovně jednoho milionu testovaných sloučenin. Uvedený koktejl se
aplikuje na nativní proteinový krystal, který si sám vybere nejvhodnější
sloučeninu. Tento proces se opakuje s jinými koktejly. Následný difrakční
experiment je plně automatizový a celý krystalografický screening trvá pouze
desítky hodin. Úlohou proteinové krystalografie je určit typ navázaného ligandu
a jeho pozici v proteinové struktuře.
Metoda
Naše metoda [2] na
lokalizaci malých molekul v protein-ligand komplexech, fázovaná
rotační-konformační-translační funkce (PRCTF) je určitou analogií metody
molekulového nahrazování (MR). Všeobecně, PRCTF je principiální nástroj na
lokalizaci molekulových fragmentů v elektronové hustotě krystalu a může být
formulována jako
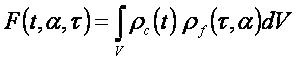 (1)
(1)
kde ρc je
elektronová hustota krystalu, definovaná pro kouli o objemu V (s poloměrem Rex)
v okolí mřížkového bodu, který přísluší translačnímu vektoru t. ρf
je elektronová hustota hledaného fragmentu. Elektronová hustota fragmentu je
funkcí konformace (vektor torzních úhlů, τ) a orientace (tři Eulerový
uhly, vektor α). Geometrický střed fragmentu je umístněn do počátku
souřadného systému. Elektronovou hustotu v kouli je možné popsat pomocí sférických
harmonických (Ylm) a sférických Besselových [gl(knlr)]
funkcí (určitá analogie atomových orbitalů, s využitím hlavního, orbitalového a
magnetického kvantového čísla) a rozvojové koeficienty je možné vypočítat ze
strukturních faktorů:
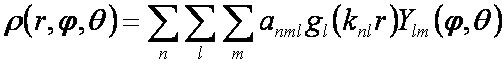 (2)
(2)
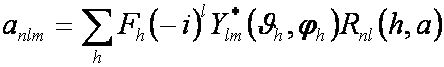 (3)
(3)
Analogicky je možné určit
rozvojové koeficienty elektronové hustoty fragmentu:
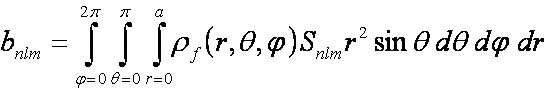 (4)
(4)
Orientaci fragmentu je
potom možné počítat pomocí FFT. Koeficienty Clmm se vypočítají z
koeficientů anlm a bnlm. Dmm jsou Wignerovy
matice pro moment hybnosti používané v kvantové mechanice.
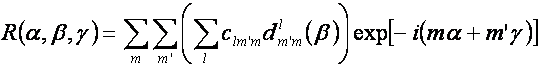 (5)
(5)
V PRCTF se na lokalizaci
fragmentů v elektronové hustotě, v skutečnosti elektronová hustota vůbec
nepoužívá. Metoda je implementována v programu NUT [2].
Pro velmi malé fragmenty,
nízké rozlišení nebo nepřesné fáze strukturních faktorů nemá překryvový
integrál (1) někdy dostatečnou schopnost odlišit správná a falešná řešení. Za
tímto účelem byl napsán malý program DOK, který počítá interakční
energii mezi ligandem a proteinem v příslušné proteinové dutině (cavity). Kombinací dvou fyzikálně odlišných přístupů je možné eliminovat
prakticky všechna nesprávná řešení.
Odkazy
1. W. Jahnke, D.A. Erlanson,
Fragment-based Approaches in drug Discovery, WILEY-VCH, Weinheim,
Germany, 2006.
2. F. Pavelcik, J.
Appl. Cryst. 39, (2006), 483-486.