Výpočetní postupy v molekulární mechanice P. Kovář, M. Veteška Univerzita Karlova v Praze, Matematicko-Fyzikální fakulta, Ke Karlovu 3, 121 16 Praha 2Kovar@karlov.mff.cuni.cz
Keywords: potential functions, molecular mechanics, minimization algorithms.
AbstractThe types of interatomic potential functions used in molecular simulations are described. Methods of calculations of non-bonded interactions are mathematically described and algorithms used in molecular mechanics are discussed.
1 ÚvodVlastnosti systémů (krystaly, molekuly), jako jsou jejich stabilita, mechanické vlastnosti, hustota, infračervená a rentgenová spektra, atd., souvisejí s jejich strukturou. Je proto vhodné vytvořit algoritmy a programy, které jsou schopny vypočítat strukturu vyšetřovaných systémů, na základě jejíž znalosti budeme pak schopni odvodit požadované vlastnosti. Molekulární simulace jsou jednou z těchto technik, které nám umožní strukturu daného systému určit. Především kvůli jednoduchosti a efektivitě pro velké struktury patří k nejrozšířenějším. Při uvažovaných velikostech struktur řádově ve stovkách až tisících atomů na jednu krystalovou buňku je nemožné v současnosti vypočítat strukturu ab-initio výpočty. Princip molekulárních simulací spočívá ve výpočtu potenciální energie pro dané uspořádání atomů (popř. atomů krystalové buňky) pomocí empirických silových polí. V tomto zjednodušeném popisu je energie systému určena započtením všech sil mezi atomy, přičemž výpočet je založen na mechanickém přístupu, tj. atomy se chovají jako mechanické oscilátory. Molekulární simulace spočívají v určení celkové potenciální energie systému a její minimalizace, kde energie je dána pomocí empiricky určených funkcí a jejich parametrů. Tyto funkce a parametry se dohromady nazývají silové pole. V silovém poli jsou výrazy pro popis vazeb mezi atomy a výrazy pro nevazebné interakce, které závisí jen na vzdálenosti mezi atomy – jedná se o elektrostatické a van der Waalsovy interakce a vodíkové můstky. Ze všech členů se pak pro konkrétní model utvoří výraz pro energii, která je funkcí souřadnic atomů v daném modelu. Celková potenciální energie systému je tedy vyjádřena jako součet vazebních Evaz a nevazebních Envaz interakcí [1, 2]: E
= Evaz + Envaz (1)
1.1 Vazebné interakceVazební člen v sobě zahrnuje členy, které charakterizují kovalentní vazbu pomocí jednoduchých analytických funkcí. Platí:
Evaz = Eb + Eθ
+ Eφ + Eδ . (2)
Eb (tzv. „bond stretch“ člen) popisuje deformaci vazebné
délky. V harmonické aproximaci představuje pružnou část vazebné síly danou
Hookovým zákonem:
![]() (3)
(3)
kde kij je silová konstanta a rij0
je ideální vazebná délka. Případná
anharmonicita potenciální energie se zohledňuje např. pomocí tzv. Morseho
potenciálu:
![]() (4)
(4)
kde D0 je vazební energie. Eθ popisuje deformaci vazebního úhlu mezi párem atomů i a k, které jsou navíc vázány k jednomu společnému atomu j (viz obr. 1):
![]() (5)
(5)
kde kijk je síla pružiny udržující vazební úhel θijk mezi atomy i, j, k na ideální hodnotě θijk0 . 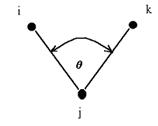
Obr. 1 Vazební úhel mezi třemi atomy. Pro vazby s úhlem blízko π tento vztah příliš nevyhovuje, proto se do vztahu zavádí vyšší mocniny 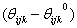 , nebo se nahrazuje různými typy kosinového rozvoje, z nichž nejjednodušší má tvar
, nebo se nahrazuje různými typy kosinového rozvoje, z nichž nejjednodušší má tvar 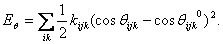 (6)
(6) Jako doplněk k Eθ slouží člen EUB (tzv. Urey-Bradley potenciál), který je v nejjednodušším tvaru dán 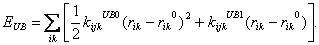 (7)
(7) je deformace úhlové torze, jež popisuje geometrii 4 atomů i, j, k, l a je charakterizována úhlem
mezi rovinami určenými polohami atomů ijk a jkl (viz obr. 2). Nejjednodušší tvar pro
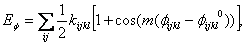 (8)
(8) kde kijkl je výška rotační bariéry okolo torzního úhlu 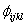 , m je periodicita potenciálové funkce a
, m je periodicita potenciálové funkce a 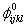 je předpokládaná rovnovážná hodnota úhlu.
je předpokládaná rovnovážná hodnota úhlu.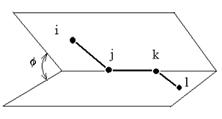
Obr. 2 Úhlová torze
Eδ je tzv. inverzní člen, který popisuje vazební geometrii 4 atomů charakterizující odchylky od planárního uspořádání (proto také zvaný „out-of-plane deformace). Podle potřeby se používají tři různé popisy:
Umbrella inverze 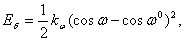 (9)
(9) kde kω je silová konstanta, ω je úhel mezi osou il a rovinou ijk a ω0 je rovnovážná hodnota tohoto úhlu. 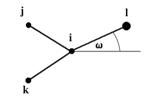
Obr. 3 Umbrella inverze
Amber inverze 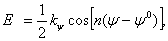 (10)
(10) kde kψ je energetická rotační bariéra, n periodicita, ψ je úhel mezi rovinami jil a kil (viz obr. 4) a ψ0 je rovnovážná hodnota tohoto úhlu. Pro planární uspořádání je n = 2 a ψ0 = 180° a pro tetraedrickou koordinaci je n = 3 a ψ0 = 120° . 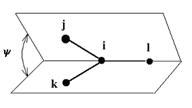
Obr. 4 Amber inverze
Charm inverze 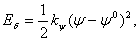 (11)
(11)kde kψ je silová konstanta, ψ je úhel mezi rovinami ijk a ljk a ψ0 je rovnovážná hodnota tohoto úhlu. 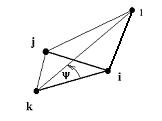
Obr. 5 Charm inverze 1.2 Nevazebné interakceNevazebné interakce Envaz zahrnují elektrostatické Eel a van der Waalsovy interakce EvdW a vodíkové vazby EHb . Pro Coulombické interakce platí 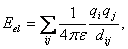 (12)
(12) kde qi a qj jsou náboje atomů i a j, dij jejich vzdálenost a ε dielektrická konstanta. Náboje jsou buď celočíselné (pro ionty), nebo parciální, reprezentující polaritu molekuly (dipól, kvadrupól, atd.). Van der Waalsovy interakce se počítají buď pomocí exponenciálního výrazu, tzv. Buckinghamova potenciálu 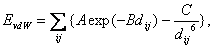 (13)
(13) kde A, B, C jsou konstanty a dij vzdálenost atomů. Další výraz pro van der Waalsovy interakce je Lennardův-Jonesův potenciál: 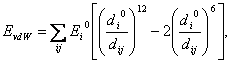 (14)
(14) kde −Ei0 je minimální vdW energie atomu, di0/2 jeho tzv. van der Waalsův poloměr a dij vzdálenost atomů. Vodíkové vazby se počítají podobným způsobem 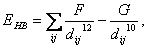 (15)
(15) kde F , G jsou konstanty a dij vzdálenost páru donor-akceptor. Omezení dosahu interakcíSložitost výpočtu nevazebných interakcí kvadraticky závisí na počtu atomů v systému, což pro větší systémy může představovat problém a je třeba učinit ořezání (cut-off), tj. stanovit mez, nad kterou se interakce mezi atomy zanedbávají nebo počítají přibližně. Zejména pak pro krystaly, kde sumace ve vztazích pro tyto interakce obsahuje nekonečný počet členů,
je nezbytné omezení a existují metody, jak tyto sumy počítat efektivním způsobem, např. Ewaldova sumace [3]. Je třeba rozlišit krátkodosahové interakce klesající rychleji než 1/d3 (např. Lennardův-Jonesův potenciál, která klesá jako 1/d6 ), které je možno zanedbat již pro vzdálenosti větší než několikanásobek interakčního minima, a dalekodosahové interakce (např. Coulombická interakce slábnoucí jako 1/d pro interakci náboj-náboj), které nelze již jednoduše pro větší vzdálenosti zanedbat. Je možno například provést přímé ořezání, kdy jsou interakce nad jistou vzdálenost ignorovány. Tato metoda ale může vést k nespojitostem v ploše energie a jejích derivacích. Tento problém se řeší přechodovými funkcemi, které vedou k hladkému přerušení interakcí. Větším problémem u této přímé metody je rozdělení dipólů, kdy jeden z pólů zůstane pod hranicí ořezu a druhý nad a vzniká tak nefyzikální monopól. Protože příspěvek k energii z interakce monopólů je o pár řádů větší než z interakce dipólů zformovaných z monopólů, takové rozdělení dipólu přináší velké chyby výpočtu energie a je třeba mu předejít. Řeší se to např. metodou ořezávání po nábojových skupinách, což jsou malé skupiny blízkých atomů, které mají společný náboj dostatečně blízko nule. Nábojovými skupinami bývají obvykle běžné chemické funkční skupiny. Pro periodické systémy jsou přesnější a efektivnější metody na výpočet nevazebných interakcí, než je metoda ořezávání. Jedná se o multiplovou metodu a především Ewaldovu sumaci. Ewaldova technika je podrobně popsána v [3]. Spočívá v rozdělení mřížové sumy na dva členy. Mřížová suma je 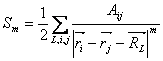 (16)
(16) kde 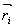 a
a 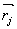 jsou bázové vektory atomů i a j a
jsou bázové vektory atomů i a j a 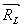 je mřížový translační vektor, přičemž sumy
je mřížový translační vektor, přičemž sumypřes i a j jdou každá přes všechny atomy v buňce. První člen z mřížové sumy vznikne jejímvynásobením konvergenční funkcí φ(r), která rychle klesá s r, druhý člen pak vynásobením 1 − φ(r):  (17)
(17) První člen velmi rychle konverguje. Druhý člen lze Fourierovou transformací převést na rychle konvergující sumu v reciproké mříži. Obě sumy lze tedy omezit na jistý počet členů. Konvergenční funkce je pro elektrostatickou energii 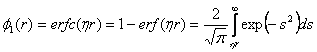 (18)
(18) a pro rozptylovou energii 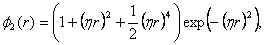 (19)
(19) kde parametr η slouží k vyrovnání rychlosti konvergence mezi sumami v reálném a reciprokém prostoru. Doba výpočtu sum závisí na zvolené velikosti omezení, tj. kolik členů z nekonečných sum se skutečně započte. 1.3 Výpočet nábojůNáboje mohou být pro jednotlivé atomy přiřazeny tzv. tabulkovou metodou, kdy příslušný parciální náboj pro daný atom v systému je odvozen například na základě kvantově-mechanických výpočtů, případně na základě experimentálních dat. K výpočtu rozložení nábojů v molekule se používá metoda Charge equilibration (QEq), která rozložení nábojů spočte v závislosti na geometrii molekuly. Metoda vychází z vyjádření energie izolovaného atomu A jako funkce jeho náboje QA. Rozvoj této energie EA(QA ) do Taylorovy řady má tvar 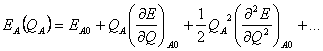 (20)
(20) Pro atomy s nábojem +1 a -1 platí: 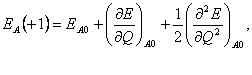 (21)
(21) 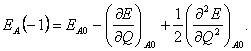 (22)
(22) Odečtením a sečtením předchozích rovnic dostaneme: 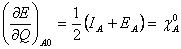 (23)
(23) 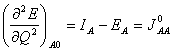 (24)
(24) kde 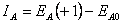 je ionizační energie (energie potřebná k odtržení elektronu z neutrálního atomu),
je ionizační energie (energie potřebná k odtržení elektronu z neutrálního atomu), 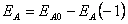 je elektronová afinita (energie uvolněná při vzniku aniontu z neutrálního atomu) a χA0 je elektronegativita atomu (schopnost daného atomu přitahovat elektrony). Pro pochopení fyzikálního smyslu (24) lze uvažovat atom s orbitalem
je elektronová afinita (energie uvolněná při vzniku aniontu z neutrálního atomu) a χA0 je elektronegativita atomu (schopnost daného atomu přitahovat elektrony). Pro pochopení fyzikálního smyslu (24) lze uvažovat atom s orbitalem 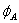 obsazeným jen jedním elektronem. Při odtržení elektronu je orbital atomu prázdný, při
obsazeným jen jedním elektronem. Při odtržení elektronu je orbital atomu prázdný, přizachycení elektronu je orbital obsazený dvěma elektrony. 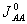 , dané rozdílem IA a EA, je tak Coulombická repulze mezi dvěma elektrony na orbitalu . Vztah pro energii izolovaného atomu (20) lze přepsat s použitím (23) a (24) na
, dané rozdílem IA a EA, je tak Coulombická repulze mezi dvěma elektrony na orbitalu . Vztah pro energii izolovaného atomu (20) lze přepsat s použitím (23) a (24) na 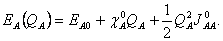 (25)
(25) Celková energie systému se získá vysčítáním přes všechny atomy: 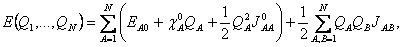 (26)
(26) kde JAB je Coulombická interakce mezi jednotkovými náboji A a B, závislá na jejich vzdálenosti. Derivace celkové energie podle náboje QA dává chemický potenciál χA, jež je funkcí nábojů: 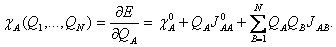 (27)
(27) 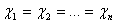 (28)
(28) Přidáním předpokladu, že součet všech nábojů na jednotlivých atomech je roven celkovému náboji na molekule 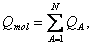 (29)
(29) se získá N rovnic pro N nábojů v rovnováze, které se pro danou strukturu řeší [4]. Pro větší modely vzdálené od rovnováhy je obvykle potřeba dříve, než se přiřadí náboje, provést částečnou minimalizaci, protože metoda QEq na deformovaných modelech může vést k výpočtu nerealistických nábojů. 2 Molekulární mechanika Molekulární mechanika se zaměřuje na statické vlastnosti řešené struktury. Hlavním cílem je optimalizovat geometrii daného modelu, tedy najít uspořádání atomů s minimální celkovou potenciální energií. Pro tuto minimalizaci se ke geometrické optimalizaci souřadnic atomů, a u periodických modelů případně i parametrů buňky, používají různé algoritmy. Další vlastnosti jsou následně počítány z této optimalizované struktury. Minimalizační algoritmyMinimalizace modelu probíhá ve dvou krocích. První spočívá ve vyhodnocení výrazu pro energii při daném uspořádání atomů. V druhém kroku je uspořádání upraveno tak, aby mělo nižší hodnotu energie. Minima může být dosaženo po jedné úpravě struktury, nebo může být potřeba tisíce iterací. To závisí na druhu algoritmu, na tvaru plochy energie a na složitosti modelu. Efektivita minimalizace se posuzuje jak časem potřebným k vyhodnocení výrazu pro energii, tak časem a počtem iterací potřebných k dosažení minima. Výraz pro energii je funkce 3N proměnných, kde N je počet atomů v modelu. Každý minimalizační algoritmus musí v tomto 3N-rozměrném prostoru určit směr k minimu a vzdálenost k minimu podél tohoto směru. Nalezený bod určuje souřadnice struktury s nižší energií. 2.1 Metoda největšího spádu (steepest-descents)Metoda největšího spádu definuje směr k minimu jako směr gradientu v daném bodě. Tento směr nevede přímo k minimu, proto se v minimu nalezeném podél tohoto směru definuje gradientem nový směr a iterativně se pokračuje. Minimum podél daného směru se vyskytuje přesně v bodě, v kterém je daný směr tečný k vrstevnicím energie. Z toho vyplývá, že nový směr určený gradientem je kolmý k předešlému. Bez ohledu na to, jakou metodou byl směr k minimu určen, se k hledání minima podél určeného směru často používá metoda, která se nazývá line search. Jedná se o jednodimenzionální minimalizaci podél směrového vektoru, při níž se minimum jednoduše uzávorkovává mezi dva body s vyšší energií. Minima je dosaženo postupnými iteracemi, v nichž se ve směru daného vektoru stále zmenšuje souvislý úsek, v němž se minimum hledá. Fakt, že následující směry jsou na sebe kolmé, zajišťuje efektivní cestu k minimu pro přibližně kvadratické výrazy pro energii. Ovšem ve skutečnosti tato metoda tím, že směr určuje nejstrmějším spádem, a tedy ne nutně přímo k minimu, nejvhodnější směr často mine, což vede k neefektivní trajektorii, která často osciluje kolem cesty k minimu. To se týká ploch energie s úzkými údolími. Tento problém je možno řešit složitějšími algoritmy, jako jsou metoda konjugovaných gradientů a Newtonova-Raphsonova metoda. Dalším problémem je náročnost hledání minima podél určeného směru. Je totiž třeba provést několik (3–10) vyhodnocení výrazu pro energii a v případě molekulární mechaniky bývá vyhodnocení výrazu tou časově nejnáročnější částí výpočtu. Tedy „line search není z tohoto hlediska moc efektivní metoda. Proto se přesné hledání minima neprovádí, ale náhodně se testují body podél prohledávaného směru a nový směr je definován v prvním bodě, v němž je energie nižší než v bodě výchozím. Výhodou je značný pokles počtu vyhodnocení výrazu během jedné iterace (na 10 %–20 %), přičemž počet iterací k dosažení minima bývá zhruba stejný jako v metodě „line search“, ačkoli trajektorie je více nepravidelná [5]. Metoda největšího spádu bývá v blízkosti minima pomalá. Ovšem vzhledem ke spoléhání se na gradienty je také velmi spolehlivá a silná, bez ohledu na to, jaký tvar má plocha energie nebo z jakého bodu výpočet začíná. Proto se tato metoda obvykle jako první v řadě používá k úvodním úpravám struktur, které jsou daleko od minima a mají velké gradienty, přičemž složitější a sofistikovanější algoritmy jsou použity následně. 2.2 Metoda konjugovaných gradientůMetoda největšího spádu konverguje pomalu v blízkosti minima, protože každý následující směr sice opraví odchylku předešlého směru od ideálního směru k minimu, ale nemůže se mu to účinně podařit vzhledem k podmínce kolmosti k předešlému směru. Metoda konjugovaných gradientů toto omezení nemá a každý následující směr je dán úpravou směru předešlého. V metodě konjugovaných gradientů je nový směrový vektor 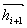 vedoucí z bodu i+1 dán připočtením gradientu
vedoucí z bodu i+1 dán připočtením gradientu 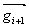 v bodě i+1 k předešlému směru
v bodě i+1 k předešlému směru 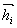 přenásobenému konstantou γi :
přenásobenému konstantou γi : 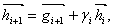 (30)
(30) kde γi může být definováno dvěma různými způsoby – v Polakově-Ribiereově metodě 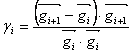 (31),
(31), a ve Fletcherově-Reevesově metodě [6] 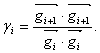 (32)
(32) Ačkoli obě metody mají podobné charakteristiky, za různých podmínek může být jedna výhodnější než druhá. Aby bylo zaručeno, že směry jsou vzájemně konjugované, je třeba minimum podél daného směru vyhledat přesně a zajistit tím, že nový gradient bude opravdu kolmý na předešlý směr. Přestože tyto výpočty tak potřebují více vyhodnocení výrazu pro energii, a proto čas na jednu iteraci může být v metodě konjugovaných gradientů delší než v metodě největšího spádu, je tato ztráta více než kompenzována mnohem efektivnější konvergencí k minimu. Je třeba dodat, že tato metoda již předpokládá uspořádání atomů natolik blízko minimu, že plocha energie je už téměř kvadratická, jinak se výpočet může stát nestabilní. Metoda konjugovaných gradientů je vhodná pro velké modely, neboť potřebuje řádově jen N údajů pro uchování předchozího směru (na rozdíl od Newtonovy-Raphsonovy metody, viz dále). Detaily o metodě v [7]. 2.3 Newtonova-Raphsonova metodaTato metoda navíc k použití gradientu, kterým se určuje směr hledání minima, použije i informaci o zakřivení plochy (druhá derivace) k výpočtu, kde se podél daného směru minimum nachází. Protože matice druhých derivací výrazu pro energii A definuje zakřivení v každém směru, přenásobením její inverze A−1(r0 ) gradientem ∇E(r0 ) ve výchozím bodě r0 lze vypočíst předpokládané minimum rmin :
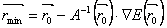 (33)
(33) Obdobně jako v předešlých metodách je třeba provádět tento postup iterativně. Nevýhoda této metody je, že výpočet matice druhých derivací je časově náročný a vzhledem k řádovým rozměrům N2 nemusí být pro velké systémy výpočetní technikou zvládnutelný. Tato metoda je ještě citlivější než metoda konjugovaných gradientů na požadavek kvadratičnosti plochy energie, v opačném případě je nestabilní. Proto se obvykle používá až jako poslední z minimalizačních algoritmů pro případ, kdy je požadována extrémní přesnost na dosažené minimum, např. pro následný výpočet vibračních frekvencí, které jsou vypočteny chybně i v případě malých zbytkových gradientů. Existují různé variace Newtonovy-Raphsonovy metody, např. kvasi-Newtonova metoda, „oříznutá Newtonova metoda, atd. Některé z nich mají výhodu, že nepotřebují k uchování N2 údajů pro matici druhých derivací. Literatura1. P. Comba, T. W. Hambley, Molecular Modeling of Inorganic Compounds. VCH, Weinheim, 1995.2. M. Veteška, Molekulární simulace ve strukturní analýze interkalátů. MFF UK Praha, 20063. N. Karasawa, W. A. Goddard III, J. Phys. Chem. 93 (1989), 7320-7327.4. A. K. Rappé, W. A. Goddard, J. Phys. Chem. 95 (1991), 3358.5. M. Levitt, S. Lifson, J. Molec. Biol. 46 (1969), 2696. R. Fletcher, C. M. Reeves, Comput. J. 7 (1964), 149.7. R. Fletcher, Practical Methods of Optimization, Vol. 1, Unconstrained Optimization. John Wiley&Sons, New York, 1980.