Structure
analysis by molecular simulation techniques.
Calculated
structures of layered silicates.
M. Pospíšil1, M. Veteška1, P. Kovář1
pospisil@karlov.mff.cuni.cz
Welcome
to the fantastic world of molecular simulations, where everything is possible,
but it is very hard work to find the truth about molecular structures which,
moreover, should correspond with the experimental data.
History and
present
Our
molecular simulations laboratory was established in 1997, when prof. Pavla
Čapková obtained a developing project. Finances from this project were used for
buying Octane computer (2x 150MHz MIPS R10000 processors) from Silicon Graphics
and package software Cerius from MSI. An interesting fact is that Octane has
been upgraded only once by two 300 MHz MIPS R12000 processors and 2GB of memory
and it is still working with IRIX64 Release 6.5 operating system. Cerius2
software has been gradually updated to Cerius2 4.5 final version.
The firm MSI was bought by Accelrys which develops their own molecular
simulation program Material Studio [MS]. Cerius was incorporated into the MS as
one module called Forcite. Cerius2 hasn’t been hereafter developed
and updated. It is necessary to say that some special features of Cerius2
software package haven’t been incorporated into Forcite and these ones are
still used in Cerius2 as complementary features. Nevertheless, MS
with its modules can cover wider area of molecular simulation problems. It is
not necessary to bye the whole software package but a buyer can buy only the
modules he demands. Nevertheless, the price of individual modules is higher
than the price for the same module in the whole software package. First of all,
a Visualiser module must be bought, that is a control interface between the user
and the modules of the program. We have several modules of MS running on 8
Intel processors PC under windows 7 operating system in our laboratory, which
we use for a research or teaching activities.
Software
The
modules in Material Studio 5.0 used in our laboratory are Forcite, Discover,
Reflex, Conformers, Blends and Synthia. There are about other 16 modules in MS.
Forcite module is used for energy calculation of the system, geometry
optimization, various kinds of dynamics, mechanical properties. Discover module
allows a parallel computing and it is used similarly like Forcite module for
energy calculations, geometry optimization, dynamics and properties analysis.
Reflex module allows solving of crystal structures from powder diffraction
pattern. Conformers module calculates the most probable conformation of
investigated molecules. Blends module is used for mixing of various components
and for description of the resultant mixture. Synthia is a module for polymer
calculation. It predicts macro properties of resultant polymers on the base of
knowledge of micro structure. This software is continuously updated together
with the computer. Moreover the computer cluster is used for calculations in
software packages like Amber, Gromacs, Gaussian, etc.
Research problems
In
our laboratory molecular simulation methods were mainly used for calculation of
various types of silicates intercalated with organic species like various
ammonium cations [1, 2], dye cations [3-5], neutral polar molecules [6], etc. Intercalated
silicates are usually in powder form. Direct diffraction methods solve the
structure of these powders with difficulties. In this case, molecular
simulations can significantly help us to describe the structures in detail. The
similar procedures are valid for anionic clays like layered double hydroxides
(LDH) with positive layer charge intercalated with benzoic acid and its
derivatives [7] and porfyrin molecules [8, 9].
Other
calculations were used for properties description of energetic materials.
Mutual interaction between molecules and intramolecular interactions have been
investigated. Dynamic calculations bring a new insight into the behaviour of energetic
materials under high pressure and high temperature [10]. Calculated result like
a time of decomposition or the energy release were compared with experimental
results like sensitivity or detonation energy. The energy characteristics from
energetic materials structures were derived and used for a possible explanation
of sensitivity behaviour [11]. Other case is a dynamic calculation of the phase
transition for liquid crystals or cocoa butter.
Teaching
The software for
molecular simulations is also permanently used for teaching of under graduated
and post graduated students. During the existence of the laboratory, 4 diploma
theses (Václav Bittner,
Miroslava Fraňová, Jana Čurdová, Marek Veteška) and 5 doctoral theses (Daniel Janeba, Miroslav Pospíšil,
Bohdan Koudelka, Jarmila Repáková, Petr Kovář) have been defended. Miroslava
Fraňová and Marek Veteška continue in the branch of the molecular simulations in their Ph.D.
theses now.
A 2 hours lecture “Molecular
simulations in chemical physics” and 1 hour seminar is regularly taught at the Faculty
of Mathematics and Physics. The students obtain a knowledge about principles,
procedures and also practical experiences with molecular calculations during
seminar. This knowledge can be amplified during 4 hour practical seminar called
“Computational experiments in molecular theory I and II” where simple mechanical and quantum chemical methods and empirical
molecular mechanics and dynamics are taught.
Future
plans
We would like to update MS software and enlarge the using of software packages installed on the computer cluster of physical section of the faculty. Moreover, we would like to increase the interconnection between classical molecular simulations and quantum calculations. We started both types of these calculation with porphyrin molecules intercalated into LDHs [9]. In the end we would like to return back to polymer calculations and we would like expand the simulations to non linear optics materials and drugs.
Figures of
solved structures
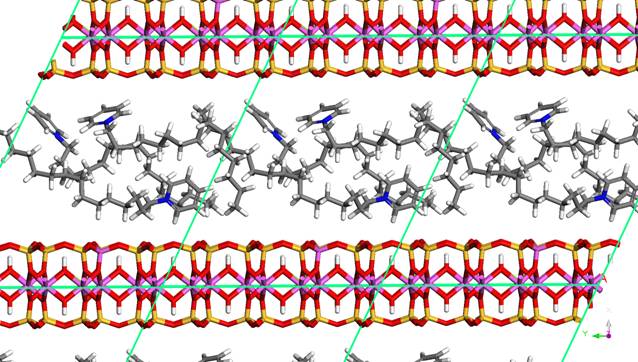
Figure 1. Montmorillonite intercalated with
cetylpyridinium cations [1]. There are 3 cations per one unit cell.
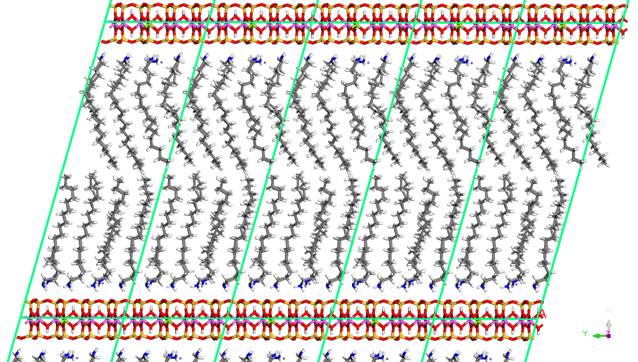
Figure 2. Montmorillonite intercalated with
neutral octadecylamine molecules [6]. There is 16 octadecylamine molecules and
3 Na cations per one unit cell.
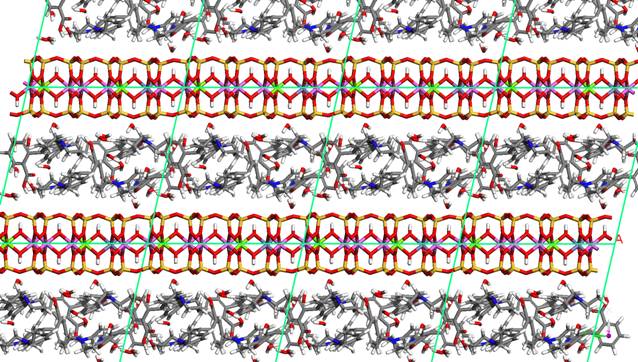
Figure 3. Montmorillonite intercalated with
rhodamine B [3]. There is 2 H dimer from 2 rhodamine B molecules and 16 water
molecules per one unit cell.
References
1. M. Pospíšil, P. Čapková, D. Měřínská,
Z. Maláč, J. Šimoník, Journal of Colloid
and Interface Science, 236, (2001),
127.
2. P. Čapková, M. Pospíšil, Z. Weiss, Journal of Molecular Modeling, 9, (2003), 195.
3. M. Pospíšil, P. Čapková, H.
Weissmannová, Z. Klika, M. Trchová, M. Chmielová, Z. Weiss, Journal of Molecular Modeling, 9, (2003), 39.
4.
P.
Čapková, P. Malý, M. Pospíšil, Z. Klika, H. Weissmannová, Z. Weiss, Journal of Colloid and Interface Science,
277, (2004), 128.
5. P. Kovář, M.
Pospíšil, P. Malý, Z. Klika, P. Čapková, P. Horáková M. Valášková, Journal of Molecular Modeling, 15, (2009), 1391.
6.
M. Pospíšil, P. Čapková, Z. Weiss, Z. Maláč, J. Šimoník, Journal of Colloid and Interface Science,
245, (2002), 126.
7.
P.
Kovář, M. Pospíšil, M. Nocchetti, P. Čapková, K. Melánová, Journal of Molecular Modeling, 13,
(2007), 937.
8.
P.
Kovář, M. Pospíšil, E. Káfuňková, K. Lang, F. Kovanda, Journal of Molecular Modeling, 16, (2010), 223.
9. E. Káfuňková,
C. Taviot-Guého, P. Bezdička, M. Klementová, P. Kovář, P. Kubát, J. Mosinger, M.
Pospíšil, K. Lang, Chemistry of Materials,
22, (2010), 2481.
10.
P.
Čapková, M. Pospíšil,
11. M. Pospíšil, P. Vávra, M.C. Concha, J.S.
Murray, P. Politzer, Journal of
Molecular Modeling, 16, (2010),
895.
Acknowledgements.
This
continuously investigation has been supported by the Czech Science Foundation
in the years 1998 to 2013 and by the Ministry of Education, Youth and Sports of
the Czech Republic (No. MSM 0021620835) and all other developing projects
allowing software packages and computer hardware up to date.