Stereochemistry of complexes of lanthanide
ions for medicinal use:
Relations between structure and function
Pavel Vojtíšek
Department of Inorganic Chemistry, Faculty of Science, Charles University, Hlavova 2030, Prague 2, 128 43, Czech Republic, pavojt@natur.cuni.cz
Polyazacycles with coordinating pendant arms are superior ligands for lanthanide ions [1,2]. Polydentate ligands, such as 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid (H4dota) and similar ligands, form thermodynamically and kinetically stable complexes even with labile metal ions such as the trivalent lanthanides [2]. Properties of such ligands have been investigated when designing magnetic resonance imaging (MRI) contrast agents [3] based on Gd3+ and diagnostic/ therapeutic radiopharmaceuticals utilising metal radionuclides such as 90Y, 64,67Cu, 111In, etc. [4]. In search for other ligands with similar or better properties than common acetate derivatives, research has also been focused on synthesis and investigation of azamacrocycles with phosphonic [5] or phosphinic [6] acids groups on pendant arms.
This lecture is focused on a stereochemistry of cyclen derivatives containing acetic acid pendant arms, methylphosphonic or methylphosphinic acid pendant arms and the similar derivatives are also included in consideration.
All of the structures show that the all H4dota-like ligands are octadentate coordinated to a lanthanide(III) ion, i.e., forming O4 and N4 planes that are parallel and have mutual angle smaller than 3°, even at cases of unsymmetrical ligands [7]. The lanthanide(III) ions lie between these planes, closer to the O4 base than to the N4 plane. All of the structures present the lanthanide(III) complexes in their twisted-square-antiprismatic configuration (TSA‘ – CN 8) with or in square-antiprismatic configuration (SA‘- CN8). The coordination shells can be fulfilled by one water molecule over the center of the O4-square; the configurations TSA with CN9 and SA with CN 9 are thus obtained.
In view of potential utilisation of the complexes as contrast agents, an important feature of these structures is the position of the water molecule. The changes in the coordination of water molecule in the the type of complexes depend on the geometry of the O4 plane as it follows from Chart 1.
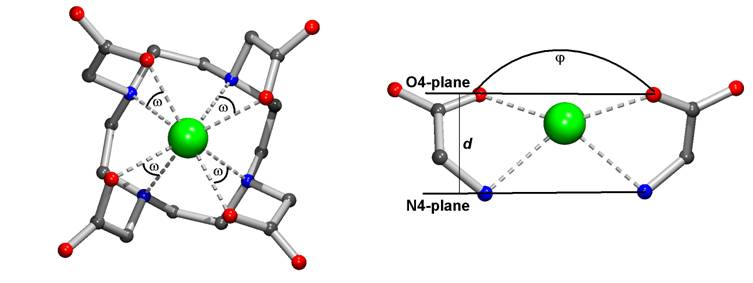
Chart 1. Schematic Drawing of the Complexes with the Depicted Structural Parameters : Twist Angle ω, Opening Angle φ, O4-plane, N4-plane, and theirs distance d.
We found [8] that for the decision on whether water molecule can or cannot be coordinated (change of CN from 8 to 9), a crucial point seems to be the O – Ln – O angle as shown in Chart 1 (angle j ). If the value of angle j is lager than 131°, coordination of water molecule is possible and the configurations TSA or SA, both with CN 9, are observed; if it is smaler, the space above the O4 plane is too small for the coordination shell expansion. The configurations TSA‘ or SA‘ with CN 8 are hence observed in these cases. It is expectable that structures with CN 9 will be more preferred for larger lanthanides [8]. But the possibility to enlarge the coordination number (CN) does not depend only on is not an ionic radius of metal ion.
The distance between N4 and O4 planes d
(see Chart 1) is another important stereochemical parameter. These values are
depicted in Fig. 1 and they are almost independent on lanthanide radius. From
the figure, it is clear that the distance found for all phosphorus derivatives
are in the range 2.63 – 2.82 Å. The distances observed for the H4dota
and H4dota-tetraamide are
split into two groups. The H4dota complexes of La(III), Ce(III), and Tm(III)
with the value of ca. 2.5 Å correspond to the TSA (La, Ce) or TSA‘ (Tm) geometry, and the other lanthanide(III)
complexes with the value of ca. 2.3 Å conform to the SA arrangement. As
shown above in the plot (Fig.1), H5do3ap
complexes (ligand with both carboxyl- and phosphorus pendant
arms) exhibit the distance of ca. 2.5
Å and, thus, follow the range of H4dota
complexes with the TSA and TSA‘ arrangements (with and without a coordinated
water molecule, respectively).
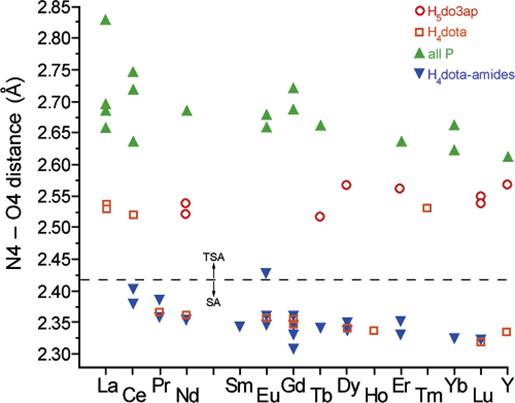
Figure 1. Dependence of distance between the O4 and N4 planes in complexes of H4dota-like ligands on the lanthanide: H5do3ap (open red circles), H4dota (open orange squares), ligands with four phosphorus atoms (full green triangles), and H4dota-amides (full blue triangles) (Fig. from [7])
We can also distinguish two main point cluster in a (non-depicted there !) plot of the distance between N4 and O4 planes as a function of twist angle ω (see Chart 1): a compact high-density cluster (ω 36°- 40°; distance of the planes near 2.35 Å) corresponding to the SA isomer and a scattered cluster (ω 22°- 32°; d 2.5 Å – 2.75 Å) containing two groups belonging to the TSA isomers. The arrangement of the points indicates that the structure of the TSA isomers is not as rigid as that of the SA isomers, being much more influenced by some other interactions. The comparison indicates that the TSA vs SA isomer preference is based on the geometry of the donor atom cavity, which is defined by the ligand, and the size of a lanthanide-(III) ion. The geometry and resulting size of the cavity in complexes of non-phosphorus H4dota-like ligands can be governed only by rotation of the pendants. This rotation leads to the different values of distances d. The SA arrangement should be preferred because of the lower repulsion of the donor atoms. Large lanthanide ions such as La(III) and Ce(III) enforce a larger cavity and thus the N4-O4 distance of 2.5 Å or higher and the formation of the TSA isomer are mostly required for their complexes. As the radius of the Ln(III) decreases, the ion moves to the N4 plane, as was documented in [8]. For non-phosphorus H4dota-like ligands, the shorter distance between the planes is adequate for the Ln(III) after Ce, and thus, the formation of the SA isomer is preferred.
In phosphonic and phosphinic acid derivatives, the pendants show a different geometry resulting from the tetrahedral arrangement of the donor groups, and in addition, the C-P and O-P bonds are longer than the C-C and O-C in the acetate pendant. Therefore, the ligands even with only one phosphonic acid group prefer longer distances O4-N4 planes and the formation of TSA isomer. The ligand cavity in TSA isomers of the small lanthanides is not rigid, as the plot twist angle ω vs distance d between N4- and O4- planes demonstrates.
Plots mentioned above for a number of Ln(III) complexes shows that the arrangement of the TSA isomers is flexible but the higher distance from O4-plane to Ln(III) can induce smaller value of openning angle φ (see Chart 1) and the coordination shell expansion is more difficult. On the other hand, the arrangement of the SA isomers is rigid but the Ln(III) ion is located nearer to O4-plane and the coordination shell expansion is favoured. Complexes of mono- and diphosphorus[9] acid derivatives make just a borderline between complexes of ligands with acetate pendants and phosphorus acid pendants.
The design of new MRI contrast agents should consider that the exclusive formation of the complexes with the more flexible TSA configuration might lead to a too-long Gd-Ow distance, thus decreasing the overall relaxivity[10]. The coordination of water molecule is easier for more rigid SA isomers but slower water exchange leads to decreasing the overall relaxivity.
The stereochemical analysis of coordination
polyhedra in lanthanide complexes with H4dota-like ligands can
demonstrate the effect of even slight structural differences on the design of
new MRI contrast agents.
[1] (a) L.F. Lindoy, Adv. Inorg. Chem. 45 (1998) 75. (b) K.P. Wainwright, Coord. Chem. Rev. 166 (1997) 35. (c) S.F. Lincoln, Coord. Chem. Rev. 166 (1997) 255.
[2] M. Meyer, V. Dahaoui-Gindrey, C. Lecomte, R. Guilard, Coord. Chem. Rev. 178–180 (1998) 1313 and refs therein.
[3] (a) D. Parker, in: J.-M. Lehn (Ed.), Comprehensive Supramolecular Chemistry, vol. 10, Pergamon Press, Oxford, 1996, pp. 487–536 and refs therein. (b) M. Botta, Eur. J. Inorg. Chem. (2000) 399.
[4] (a) C.J. Anderson, M.J. Welch, Chem. Rev. 99 (1999) 2219. (b) W.A. Volkert, T.J. Hoffmann, Chem. Rev. 99 (1999) 2269. (c) D.E. Reichert, J.S. Lewis, C.J. Anderson, Coord. Chem. Rev. 184 (1999) 3.
[5] J. Ren, A. D. Sherry, Inorg. Chim. Acta 246 (1996) 331.
[6] J. Huskens, A.D. Sherry, J. Am. Chem. Soc. 118 (1996) 4396 and refs. therein.
[7] P. Vojtíšek, P. Cígler, J. Kotek, P. Hermann, I. Lukeš, Inorg. Chem. 44 (2005) 5591–5599
[8] (a) I. Lukeš, J. Kotek, P. Vojtíšek, P. Hermann, Coord. Chem. Rev. 287 (2001) 216–217. (b) J. Klimentová, P. Vojtíšek, J. Mol. Struct. 826 (2007) 82–88.
[9] M. P. C. Campello et al. prepared to
publication.
[10] L. Burai, É. Tóth, G. Moreau, A.
Sour, R. Scopelliti, A.E. Merbach, Chem. Eur. J. 9 (2003) 1394.
Inorg. Chem. 2005, 44, 55915599