Molecular simulations of Zn-Al Layered Double Hydroxide intercalated with porphyrin anions.
P. Kovář1, M. Pospíšil1, K. Lang2
1Charles University in Prague, Faculty of Mathematics and Physics, Ke Karlovu 3, 121 16 Prague, Czech Rebublic
2Institute of Inorganic Chemistry, v.v.i., Academy of Sciences of the Czech Republic, Řež 250 68, Czech Republic
Kovar@karlov.mff.cuni.cz
Layered Double
Hydroxides (LDHs) belong to the inorganic layered solids consisted of the rigid
layers containing two kinds of metallic atoms and the interlayers containing
exchangeable compensating anions and water molecules. These materials are
attractive due to their simple modification and they are used in many branches
like catalysis, precursors for chemical reactions, drug delivery,
decontamination of water, soils, etc. The intercalation of porphyrins into
Layered Double Hydroxides based on ion exchange plays an important role in
designing of new materials with optical properties, which can be used as
photofunctional units [1]. Advanced methods of preparation of the sample allow
us to obtain well crystallized samples and their structure analysis can provide
deeper structural details. Zn(II)-5,10,15,20-tetrakis(4-sulfonatophenyl)
porphyrin – (ZnTPPS) shown in Fig. 1 was intercalated into Zn2-Al
LDH and a high crystallinity was achieved by the coprecipitation procedures
followed by a post-synthesis hydrothermal treatment.
Molecular simulations and quantum
chemistry calculations combined with X-ray diffraction, thermogravimetry and
electron density measurements were used in the structure analysis. The geometry
and dimensions of ZnTPPS were optimized by the quantum-chemistry computational
program Turbomole v5.9 using the RI-DFT method with B-P86 functional [2]. The
optimized models of porphyrins were subsequently used in the molecular
simulations. The cell parameters were determined from experimental XRD patterns:
a = b = 3.064 Å. 96 cells were linked to obtain the layer [Zn64Al32(OH)192]32+
with the lattice parameters: A = 49.024 Ǻ and B = 18.384 Ǻ.
The basal spacing in initial models was equal to the value of the experimental basal
spacing (23.05 Å) obtained from the experimental XRD data. The estimated loading
of ZnTPPS anions in the interlayer space was over 90% of anion exchange
capacity (AEC). It was approximated by structural models with the 100% loading of
AEC. The initial models contained 4 water molecules per [Zn4Al2(OH)12]2+.
The composition of initial structure models was [Zn192Al96(OH)576][(ZnTPPS)24·192
H2O] and a set of models with various
orientations of guest anions with respect to the host layers and with respect
to each other was created. The minimization of the initial models was carried
out in the Universal force field [3], the electrostatic energy was calculated
by Ewald summation method [4] and the van der
Waals energy was calculated by Lennard-Jones potential [5]. The space group was
P1 and the porphyrin pyrroles were kept in their geometry as it was obtained by
ab-initio calculations. The models were minimized iteratively in two steps,
with fixed and variable cell parameters to obtain a good estimation of the orientation
of the guest with respect to the host layers and a good agreement with the
experimental basal spacing. The minimized models were refined by quench
dynamics in an NVT (constant number of atoms, constant volume and constant
temperature) statistical ensemble at a temperature of 300 K. One dynamics step
was 0.001 ps and 200 ps of dynamics were carried out.
The results are summarized in Figs.
2 and 3. Fig. 2 shows the calculated and experimental powder XRD patterns with
basal diffraction lines characterizing the interlayer arrangement and are
compared for 2θ from 3 to 25°. The
arrangement of the guests in the interlayer space corresponding to the
calculated powder XRD pattern is shown in Fig. 3. The porphyrin anions are horizontally shifted, the horizontal
shift ranges from one third to one half of the porphyrin diameter and the
guests nearly homogeneously occupy the interlayer space. The low
intensity peaks in the calculated XRD especially between 5 and 6° and between
16 and 18° are caused by the forced periodicity of central zinc atoms of ZnTPPS.
It indicates that in the real sample one can expect no order of the guests in
the interlayer space. The ZnTPPS porphyrin planes exhibit a tilted orientation
with respect to the normal. The average calculated value of tilted angle is of
14 °.
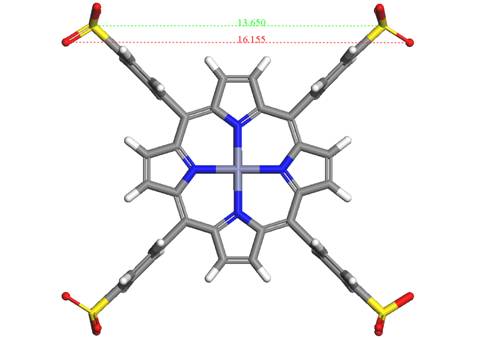
Figure 1. Molecular structure of ZnTPPS and its
dimensions.
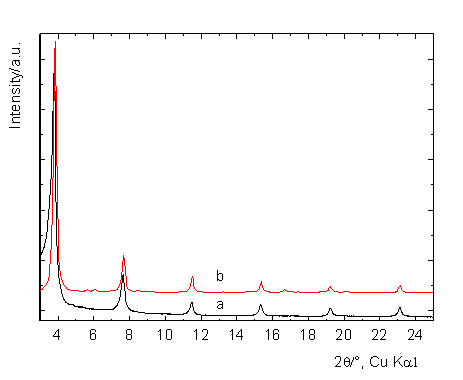
Figure 2. Experimental (a) and calculated (b) XRD basal
diffractions of Zn2Al/ZnTPPS intercalate.
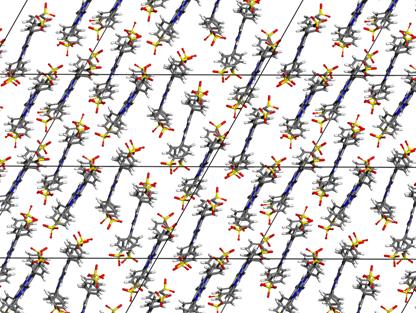
Figure 3. Top view of the linked supercells on the arrangement of the guests in the interlayer space.
1. K. Lang, P.
Bezdička, J.L.
Bourleande, J. Hernando, I. Jirka, E. Káfuňková, F. Kovanda, P. Kubát, J.
Mosinger,
D.M. Wágnerová, Chem. Mater., 19,
(2007), 3822.
2. R. Ahlrichs, M. Bär, M. Häser, H. Horn, C. Kölmel, Chem. Phys. Lett.,
162, (1989), 165.
3. A.K. Rappé, C.J. Casewit, K.S. Colwell, W.A. Goddard III, W.M. Skiff, J. Am. Chem. Soc., 114, (1992), 10024.
4. N. Karasawa,
W.A. Goddard, J. Phys. Chem., 93, (1989), 7320.
5. J.E. Lennard-Jones, Proc. Royal Soc. of London, 109, (1925), 584.
Acknowledgements.
The work was supported by GAČR 205/08/0869 and by MSM 0021620835.