Molecular modelling of Layered Double Hydroxide intercalated with benzoate
Petr Kovář1 and Pavla Čapková
1Faculty
of Mathematics and Physics,
kovar@karlov.mff.cuni.cz
Keywords: molecular modelling, structure analysis, Layered Double Hydroxide, benzoate
Abstract
The structure of Zn4Al2 Layered Double Hydroxide (LDH) intercalated with benzencarboxylate (C6H5COO-) was solved by methods of molecular modelling. Molecular modelling using empirical force field was carried out in Cerius2 modelling environment. According to the comparison of total crystal energy of optimized structure models with different geometry it was found out that benzoate anions are almost perpendicular to LDH layers, anchored to OH groups of the host layers via hydrogen bonds. Mutual orientation of benzoate ring planes in the interlayer space keeps parquet arrangement. Water molecules are roughly arranged in planes adjacent to host layers together with COO- groups.
Introduction
The object of molecular
modelling is the generation, manipulation and representation of realistic
three-dimensional molecular structures, or description of a system of
interacting molecules with the purpose of understanding the physiochemical
properties and macroscopic phenomena at the molecular level. In the past ab -
initio calculations have been extensively used to characterize the structure
and properties of a large variety of molecules [1]. Since the requirements on
the size of the investigated system are growing all the time the ab - initio
calculations remain slow to be used in realistic modelling of important systems
such as catalysts, biomolecules, polymers, etc. Nevertheless the ab - initio
calculations remain extremely useful in the determination of force field
parameters being used in molecular modeling [1] and for systems containing tens
or hundreds of atoms. Molecular modeling plays an important role in everyday
research in medical, natural and engineering sciences [2]. In some cases force
fields calculations can provide answers that are as accurate as even the
highest-level quantum mechanical calculations, in a fraction of the computer
time.
The important role by using
molecular modelling is combination with experimental data such as IR
spectroscopy, X-ray powder diffraction or statistical thermodynamic quantities
in case of molecular dynamics simulations to verify the models obtained from
calculations. A ten year - old history of molecular modelling at the Faculty of
Mathematics and Physics of Charles University has carried many important
results concerning the structure analysis and connection of the structure and
physical and chemical properties of intercalated clays by organic molecules.
Since these structures mostly exhibit a certain degree of disorder the classical
structure analysis based on X - ray diffraction fails and molecular modeling is
an appropriate tool how to reveal the character of this disorder and so wholly
clear up the structure. In this paper we report structure analysis of Zn4Al2
Layered Double Hydroxide intercalated with benzoate by methods of molecular
modelling related to diffraction.
Molecular modelling
Molecular modelling using
empirical force field was carried out in Cerius2 modelling
environment. The first stage was a construction of Zn4Al2
host framework. Unit cell of the host structure is trilayer, the space group is
R-3m with triclinic cell and lattice parameters a = 3.076
Å, c = 23.20 Å, α = 90°, β = 90°, γ
= 120°. The Al and Zn atoms in the host layers were randomly distributed so
that the composition of the host structure corresponded to the experimental
one. The measured value of basal spacing was dexp = 15.3
Å, thus the interlayer distance in the initial model was set to this
value. To investigate the arrangement of guest molecules in the interlayer
space a P1 superlattice was created with the dimensions 4a x 6a x
1c where the c = 3dexp
= 45.9 Å. The charge of this trilayer supercell was + 24 el. That means
24 benzoate anions that were created in 3-D Sketcher module were placed
into the interlayer space to compensate the charge of the layer, i.e. the
supercell consisted of 3 host layers and 3 guest layers. The composition of the
structure model was [Zn16Al8(OH)12] [C6H5COO-]8
* 24 H2O. The amount of water was obtained from
thermogravimetric measurements.
We built a series of initial
models with various positions and orientations of guests with respect to the
layers:
1/ Benzoate ring planes
parallel to the host layers in bilayer or trilayer guest arrangement in the
interlayer space.
2/ Benzoate ring planes tilted
to the host layers in bilayer guest arrangement in the interlayer space.
3/Benzoate ring planes
perpendicular to the host layers and with various mutual orientations and various
positions of COO- groups with respect to OH groups on the host
structure.
Energy minimization was
carried out in Universal force field [3]. The electrostatic energy was
calculated by Ewald summation method, van der Waals energy was expressed by
Lenard - Jones potential [4]. The minimization of the total crystal energy was
carried out in the Minimizer module according to the following strategy:
All the host layers in the
supercell were kept as rigid units during energy minimization. Variable
parameters were: c, α, β (It enabled to optimize
the mutual positions of the layers.) and all atomic positions in guest layers.
The minimization was carried out by Modified Newton procedure. The
calculated structure models were sorted according to the minimum of total
crystal energy.
The work concerning the
similarity of experimental and calculated diffraction patterns is accepted in
Journal of Molecular Modelling. A good agreement has been achieved between
modelling and experiment.
Results
Results of modelling led to
the conclusion concerning the orientation and position of the benzoate anions
and water molecules in the interlayer space. Table 1 shows total crystal energy
and basal spacing d of selected optimized models with various arrangement
of guest anions in the interlayer space (trilayer parallel, tilted,
perpendicular with a slight disorder (perp.disordered) and perpendicular
ordered arrangement (perp.ordered)). These types of structure models are
illustrated in the figures 1 - 3. Hydrogen bonds are represented by broken
lines. In the first structure model COO- groups are anchored to the
OH groups of the host layer. Since the phenyl rings are hydrophobic and the
host layers itself is hydrophilic benzoate rings of guest anions do not adopt
exactly parallel orientation with respect to the host layers but they exhibit a
slight tilted orientation. The second model with tilted orientation is shown in
the figure 2. The benzoate planes are not regularly organized with respect to
each other, the mutual departure of the ring planes is mostly about 20° but in
some cases it can exhibit a higher value. One can see from the table 1, that
the most convenient model according to the minimum of total crystal energy is
the one with ordered perpendicular arrangement of the guest anions with respect
to the host layers. One can see in the figure 3 that benzoate exhibit a slight
departure of its long axis from the perpendicular orientation in the range of
10 degrees. COO- groups are anchored to OH groups of the LDH layer
via hydrogen bonds. The detailed view of orientation of COO- groups
with respect to the OH groups is shown in the figure 4.
The structure exhibits a high
degree of order of the guest anions in the interlayer space. The benzoate
planes keep parquet arrangement as it is shown in the figure 5. Molecular
modelling revealed two types of structure models with two different
orientations of benzoate anions that keep parquet arrangement. The first type
is shown in the figure 3 where two neighboring benzoates exhibit the same
orientation of COO- groups. The second one shown in the figure 6
exhibits opposite orientations of COO- groups with respect to each
other. It is seen in the figure 5 that
COO- groups can freely rotate around the long axis of the guest
anions. The energy minimization procedure showed that water molecules are not
randomly distributed in the interlayer space but they are aggregated in planes
adjacent to host layers coinciding with COO- planes in case of
perpendicular arrangement of guests with respect to the layers.
Table 1. Comparing of total crystal energy of
optimized models with various
arrangement of
guests (parallel orientation,
tilted, perpendicular
with a slight disorder
(perp.disordered) and per-
pendicular ordered arrangement)
|
Model |
Etotal / kcal |
d / Å |
fig.No. |
|
parallel |
-4750 |
15,57 |
1 |
|
tilted |
-3081 |
15,10 |
2 |
|
perp. disorered |
-11757 |
15,20 |
3 |
|
perp. ordered |
-13227 |
15,20 |
3 |
Conclusion
This work shows that molecular
modelling using empirical force field is a powerful instrument in structure
analysis in case of hydrotalcite - like compounds. The energy minimization
revealed that the most probable structure model, i.e. model corresponding to
the energy minimum, exhibits a high degree of order of guest anions in the
interlayer space that are oriented perpendicular to the host layers. The basal
spacing d = 15.2 Å of this structure model is in good agreement with
experimental basal spacing dexp
= 15.3Å. This shows that Universal
forcefield used in these calculations is able to well describe these
structures. One can see that in this case molecular modelling is a sufficient
instrument for structure solving based only on the comparing of the total
crystal energy of optimized models obtained from a series of models with
various starting geometry. Resulting model was also in good agreement with
experimental data.
References
[1] M. A. C. Nascimento, Molecular Modeling.
[2] T. F. Kumosinski, M. N. Liebman, Molecular Modeling.
[3] Cerius2 User guide, Forcefield - Based Simulations.
[4] J.E. Lennard-Jones, Proc. Of the Royal Society of London,
series A, 109 No. 752, (1925), 584
Acknowledgements.
This
work was supported by grant MSM
0021620835
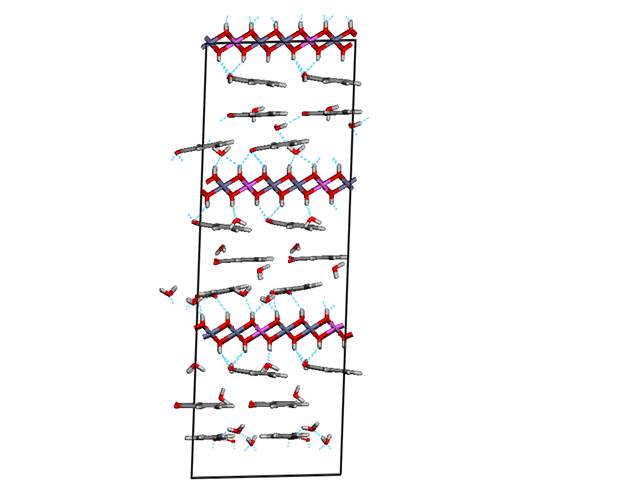
Figure 1. Structure model with
parallel orientation of guest anions with respect to the host layers
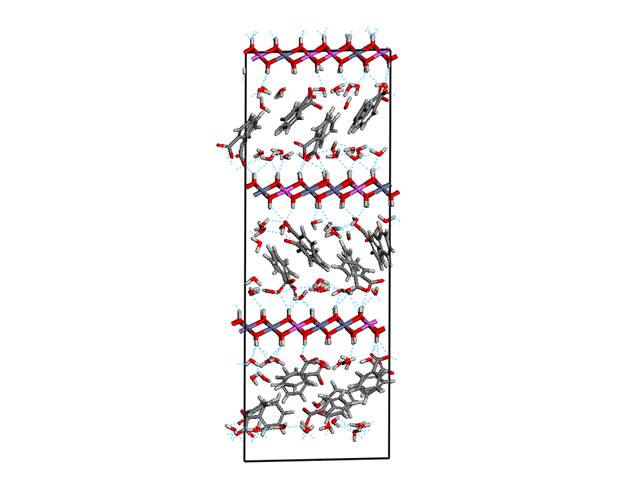
Figure 2. Structure model with
tilted orientation of guest anions with respect to the host layers
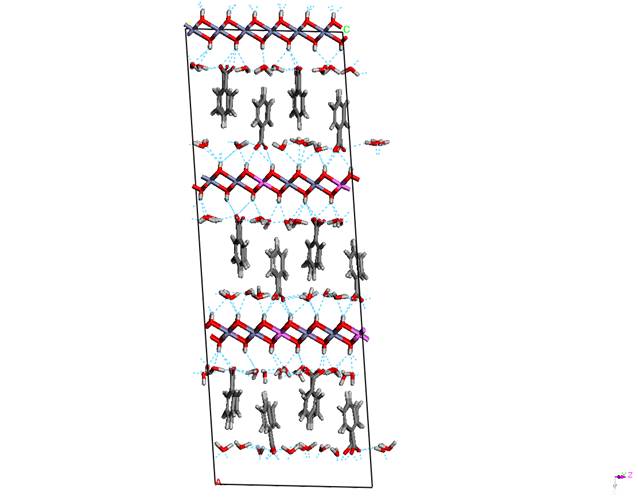
Figure 3. Structure model with
perpendicular orientation of guest anions with respect to the host layers
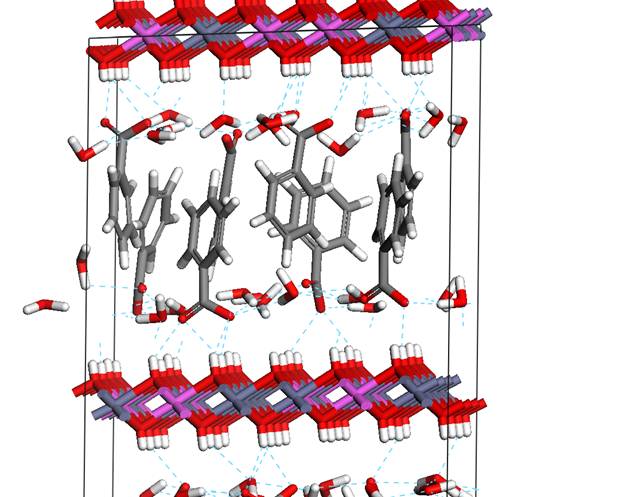
Figure 4. Detailed view of
orientation of COO- groups
with respect to OH groups
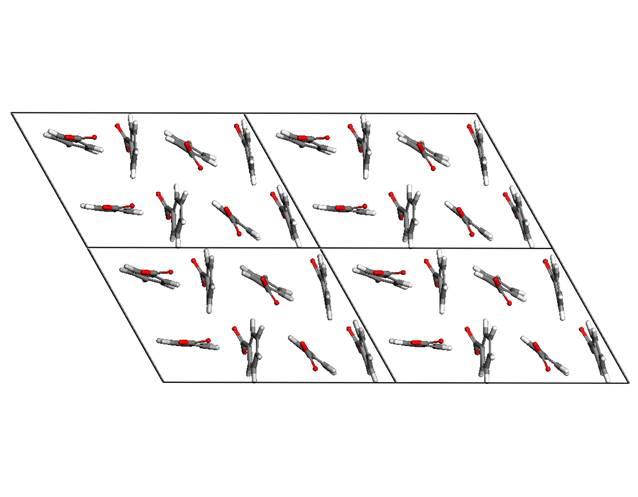
Figure 5. Parquet arrangement
of benzoate ring planes
Figure 6. Opposite orientation
of COO- groups