A Complex of HIV-1 Protease and Phenylnorstatine
Inhibitor Analyzed at Atomic Resolution
J. Brynda1, P. Rezacova1,
M. Fabry1, M. Horejsi1, R. Stouracova1, M.
Soucek2, M. Hradilek2, M. Lepsik2, J. Konvalinka2 and J. Sedlacek1
1Institute of Molecular Genetics, Academy of Sciences of the Czech Republic, Flemingovo nám. 2, 16637 Prague 6
2Institute of Organic Chemistry and Biochemistry, Academy of Sciences of the Czech Republic, Flemingovo nám. 2, 16610 Prague 6
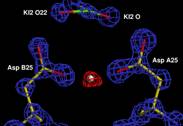 The x-ray structure of a complex of HIV-1 protease (PR) with a
phenylnorstatine inhibitor Z‑Pns‑Phe‑Glu‑Glu‑NH2
has been determined at 1.03 Å, the highest resolution so far
reported for any HIV PR complex. The inhibitor shows subnanomolar Ki values for
both the wild‑type PR and the variant representing one of the most common
mutations linked to resistance development. The structure comprising the
phenylnorstatine moiety of (2R,3S)
chirality displays a unique pattern of hydrogen bonding to the two catalytic
aspartate residues. This high resolution allows to assess the donor/acceptor
relations of this hydrogen bonding, and to indicate a proton shared by the two
catalytic residues.
The x-ray structure of a complex of HIV-1 protease (PR) with a
phenylnorstatine inhibitor Z‑Pns‑Phe‑Glu‑Glu‑NH2
has been determined at 1.03 Å, the highest resolution so far
reported for any HIV PR complex. The inhibitor shows subnanomolar Ki values for
both the wild‑type PR and the variant representing one of the most common
mutations linked to resistance development. The structure comprising the
phenylnorstatine moiety of (2R,3S)
chirality displays a unique pattern of hydrogen bonding to the two catalytic
aspartate residues. This high resolution allows to assess the donor/acceptor
relations of this hydrogen bonding, and to indicate a proton shared by the two
catalytic residues.
A structure-aided design of HIV PR inhibitors has led to a class of drugs useful in clinical anti-HIV intervention [see ref. 1 for review]. Nevertheless, mutational development of HIV PR drug resistance presents a major medical complication.
A combinatorial chemistry approach2 yielded a series of novel pluripotent HIV PR inhibitors having a picomolar range of their Ki values for the wild‑type HIV PR as well as various degrees of insensitivity of their inhibitory potency to HIV PR variants with mutations in positions 48, 82, 84 and 90, often found in drug-resistant PR strains3. In this work, the structure of wild-type HIV PR, complexed with one of these inhibitors, Z‑Pns‑Phe‑Glu‑Glu‑NH2 (Z, benzyloxycarbonyl; Pns, phenylnorstatine, (2R,3S)-3-amino-2-hydroxy-4-phenylbutanoic acid, termed hereafter KI2), is described. The phenylnorstatine group, an untypical inhibitor moiety, served the purpose to investigate the potential of replacement of amino acid residues with larger groups (the "main chain" between the aromatic groups occupying S1' and S1 pockets is longer by one carbonyl group, as compared to common, e.g., hydroxyethylene or hydroxyethylamine isosteres). This compound inhibits the Val82Ala mutant of HIV PR with a Ki value 0.11 nM, while the wild-type HIV PR inhibition has Ki = 0.18 nM3.
1. Wlodawer, A.; Vondrasek, J. Inhibitors of HIV-1 Protease: A major success of structure-assisted drug design. Anu. Rev. Biophys. Biomol. Struct. 1998, 27, 249-284.
2. Houghten, R. A.; Pinilla, C.; Blondelle, S. E.; Appel, J. R.; Dooley, C.T.; Cuervo, J. H. Generation and use of synthetic peptide combinatorial libraries for basic research and drug discovery. Nature 1991, 354, 84-86.
3. Rinnova, M.; Hradilek, M.; Barinka, C.; Weber, J.; Soucek, M.; Vondrasek, J.; Klimkait, T.; Konvalinka, . A picomolar inhibitor of resistant strains of human immunodeficiency virus protease identified by a combinatorial approach. J. Arch. Biochem. Biophys. 2000, 382, 22-30.
Acknowledgement
The
authors wish to thank the staff of ESRF in Grenoble, beamline ID14.