NMR KRYSTALOGRAFIE: Studium
struktury materiálů pomocí moderních NMR metod
J. Brus
Ústav
makromolekulární chemie AV ČR, Heyrovského nám. 2, 162 06 Praha 6 – Petřiny
 NMR spektroskopie pevného stavu je v
současné době rychle se vyvíjející oblast strukturní analýzy. Nedávný rozvoj
nových metodik a základních elektronických součástí NMR spektrometrů vedl
k tomu, že dosažené rozlišení a selektivita NMR experimentů v pevné
fázi umožňuje velmi přesně popisovat strukturu a vnitřní pohyblivost širokého
spektra látek a systémů (od velice tvrdých a rigidních skel, organických i
anorganických krystalů, přes syntetické polymery až po velice měkké a pohyblivé
gely polypeptidů či proteinů). Díky tomu je možné popsat strukturu a
tří-dimenzionální uspořádání i u takových látek, které jen velmi neochotně
poskytují krystaly vhodné k rtg. difrakci. Navíc je řada NMR parametrů
citlivá na rychlost a amplitudu vnitřních pohybů, a tak právě NMR spektroskopie
pevného stavu podává komplexní informace o vnitřní struktuře a uspořádání
hmoty. Proto můžeme NMR spektroskopii pevného stavu směle považovat za metodu
komplementární k rentgenové difrakci. Cílem NMR spektroskopiků, ale není
zcela nahradit difrakční techniky, ale především doplnit chybějící údaje k
úplnému popisu struktury a dynamiky krystalických a vysoce organizovaných
systémů. Přestože NMR spektroskopie pevného stavu tedy není alternativou
difrakčním technikám při určování úplné třídimenzionální struktury a uspořádání
molekul v krystalických materiálech, poskytuje tato metoda významné
krystalografické informace. Nejen díky tomu se tak NMR spektroskopie pevného
stavu stala významnou součástí charakterizace farmaceuticky aktivních substancí
a od roku 1997 je doporučována i hlavním regulátorem trhu - FDA (Food and Drug
Administration) [1].
NMR spektroskopie pevného stavu je v
současné době rychle se vyvíjející oblast strukturní analýzy. Nedávný rozvoj
nových metodik a základních elektronických součástí NMR spektrometrů vedl
k tomu, že dosažené rozlišení a selektivita NMR experimentů v pevné
fázi umožňuje velmi přesně popisovat strukturu a vnitřní pohyblivost širokého
spektra látek a systémů (od velice tvrdých a rigidních skel, organických i
anorganických krystalů, přes syntetické polymery až po velice měkké a pohyblivé
gely polypeptidů či proteinů). Díky tomu je možné popsat strukturu a
tří-dimenzionální uspořádání i u takových látek, které jen velmi neochotně
poskytují krystaly vhodné k rtg. difrakci. Navíc je řada NMR parametrů
citlivá na rychlost a amplitudu vnitřních pohybů, a tak právě NMR spektroskopie
pevného stavu podává komplexní informace o vnitřní struktuře a uspořádání
hmoty. Proto můžeme NMR spektroskopii pevného stavu směle považovat za metodu
komplementární k rentgenové difrakci. Cílem NMR spektroskopiků, ale není
zcela nahradit difrakční techniky, ale především doplnit chybějící údaje k
úplnému popisu struktury a dynamiky krystalických a vysoce organizovaných
systémů. Přestože NMR spektroskopie pevného stavu tedy není alternativou
difrakčním technikám při určování úplné třídimenzionální struktury a uspořádání
molekul v krystalických materiálech, poskytuje tato metoda významné
krystalografické informace. Nejen díky tomu se tak NMR spektroskopie pevného
stavu stala významnou součástí charakterizace farmaceuticky aktivních substancí
a od roku 1997 je doporučována i hlavním regulátorem trhu - FDA (Food and Drug
Administration) [1].
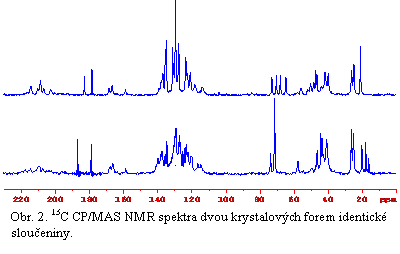
Aplikovatelnost této metody ve farmacii je však podmíněna možností poskytovat požadované informace v relativně krátkém čase a možností analyzovat látky v přirozeném izotopickém zastoupení. Z toho plyne, že základním nositelem strukturních informací je především chemický posun. Ten velice citlivě reaguje i na nepatrné změny v rozložení hustoty elektronů v okolí detekovaných jader. Proto je prvním krokem při určování strukturních parametrů měření chemického posunu rozličných jader (1H, 13C, 19F, 15N, 31P) a nalezení základních vztahů a zákonitostí mezi tímto parametrem a strukturou hmoty. V tom nejjednodušším přiblížení NMR chemický posun slouží k rozlišení a identifikaci jednotlivých polymorfů, solvatomorfů, či hydrátů. Zajímavou možností je také schopnost identifikace nestechiometických hydrátů a lokalizace molekul vody v krystalových kanálech. Měření anizotropie chemického posunu (CSA) pak může podat cenné informace o koordinaci některých těžkých atomů. V neposlední řadě pak analýza NMR spekter krystalických látek může vést k získání takových parametrů jako je určení krystalografické asymetrické jednotky nebo prostorové grupy. Tyto informace spolu s pochopením povahy vodíkových vazeb poutající molekuly do třídimenzionální struktury a lokalizace odpovídajícího vodíkového atomu následně umožní získat významná geometrická omezení. Na rozdíl od difrakčních technik dokáže NMR spektroskopie velmi přesně lokalizovat pozici vodíkových atomů. To potom vede k podstatně snadnější interpretaci především rtg. práškových dat a rekonstrukci a optimalizaci úplné 3D struktury. NMR spektroskopie také významně přispívá k rozlišení a výzkumu statického či dynamického neuspořádání krystalové struktury a nedávno rozvinuté techniky umožňují poměrně přesně popsat geometrii nebo amplitudu lokálních segmentálních pohybů, jejichž korelační čas je kratší než 40 ms.
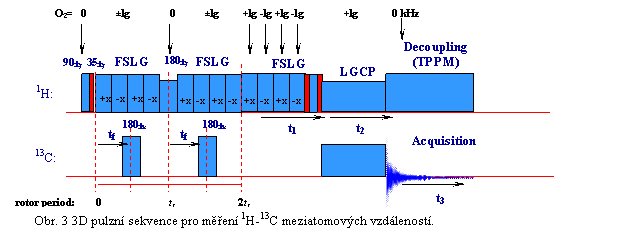
NMR spektroskopie pevného stavu se v současné době ale již stala natolik výkonnou sondou do elektronového okolí daného jádra, že její jedinečná selektivita, umožňuje za jistých podmínek přesně stanovit mezijaderné vzdálenosti až do 5 A. Faktem ale je, že v přirozeném izotopickém zastoupení můžeme měřit pouze takové meziatomové vzdálenosti, kdy alespoň jedním z partnerů je izotop s vysokým přírodním zastoupením. To jsou především jádra 1H, 19F nebo 31P. A tak nejčastěji měříme meziatomové vzdálenosti 1H-1H, 1H-13C, 1H-15N, 31P-13C atp. Podstatou použitých experimentálních postupů je přesné měření vybraných jaderných dipolárních interakcí, ve kterých jsou hledané strukturní informace uloženy. To vyžaduje nalezení speciálních podmínek a provedení více-dimenzionálních experimentů (tří až čtyř-dimenzionálních, viz. Obr. 3), které umožní selektivní detekci dipolárních spekter pro jednotlivé efektivně „izolované“ spinové páry.
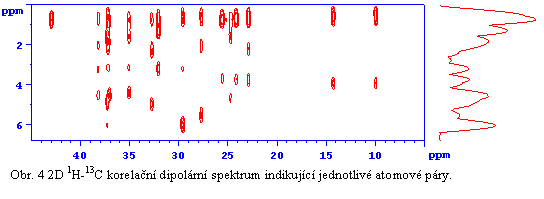
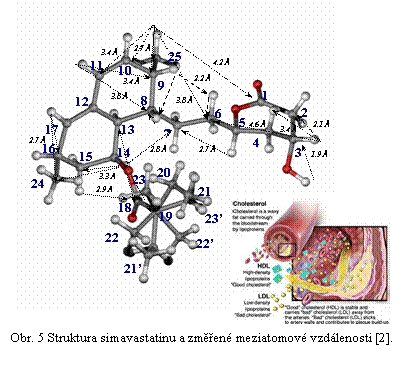
Z analýzy a simulace dipolárních spekter (viz. Obr. 4) pak lze získat požadované strukturní parametry a z těch nakonec určit konformaci a strukturu systémů s vysokým stupněm uspořádání. Přesnost měření meziatomých vzdáleností je ± 2 pm pro vzdálenosti do 200 pm a ± 5 pm pro délky do 400 pm. Úplné vyřešení 3D struktury a konformace molekul v pevné fázi, ale zdaleka není rutinní technikou. Naopak vyžaduje maximální možnou selektivitu a rozlišení, potlačení nežádoucích koherencí při zachování vysoké účinnosti excitace („recouplingu“) dipolárních interakcí a co možná nejmenší časovou náročnost.
[1] Q6a Specifications: Test Procedures and Acceptance Criteria for New Drug Substance and New Drug Products: Chemical Substance, U.S. Goverment Federal Register 1997, 62(227) 62890-62910.
[2] J.
Brus, A. Jegorov, J. Phys. Chem. A., 108 (2004) 3955-3964.