Nábojové hustoty a topologická analýza
Miroslav Šlouf, ÚMCH AV ČR, Heyrovského nám. 2, 16206 Praha 6, slouf@imc.cas.cz
Osnova
1. Úvod
2. Nábojové hustoty
2.1. Nábojové hustoty a XRD
2.1.1. Základní teorie
2.1.2. Praktické výpočty
2.2. Nábojové hustoty a QM
2.2.1. Základní teorie
2.2.2. Praktické výpočty
3. Topologická analýza nábojových hustot
3.1. Základní pojmy
3.2. Praktické výpočty v XRD
3.3. Praktické výpočty v QM
4. Závěr
1. Úvod
Rozložení elektronů v krystalu je popsáno funkcí tří proměnných r(x,y,z), která se nazývá elektronová hustota (electron density) nebo nábojová hustota (charge density), přičemž proměnné x, y, z jsou frakční souřadnice. Funkci r(x,y,z) je možno získat z experimentu pomocí rentgenostrukturní analýzy monokrystalů (single-crystal X-ray diffraction; XRD) i z teoretických výpočtů pomocí kvantové mechaniky (quantum mechanics, QM). Elektronová hustota, potažmo funkce r(x,y,z), má určité charakteristické rysy, např. není nikde záporná a nabývá maximálních hodnot v blízkosti atomových jader; studiem těchto a dalších rysů funkce r(x,y,z) se zabývá topologická analýza (topological analysis; TA) nebo podrobněji topologická analýza elektronové (či nábojové) hustoty. TA dokáže pomocí přesně definovaných čísel popsat pojmy jako atomový náboj, iontovost a excentricita vazby, dvojný charakter vazby a podobně.
2. Nábojové hustoty
2.1. Nábojové hustoty a XRD
2.1.1. Základní teorie
V rentgenostrukturní analýze měříme intenzity záření Ihkl difraktovaného na krystalových rovinách (hkl), které jsou úměrné čtvercům absolutních hodnot strukturních faktorů Fhkl:
![]() . (1)
. (1)
Strukturní faktory jsou obecně komplexní čísla, což můžeme vyjádřit pomocí rovnice:
![]() , (2)
, (2)
kde jhkl jsou fáze a |Fhkl| amplitudy strukturních faktorů. Skutečnost, že experiment poskytuje pouze amplitudy |Fhkl|, zatímco k následnému určení krystalové struktury jsou nezbytné hodnoty Fhkl se nazývá fázový problém rentgenostrukturní analýzy, jeho řešení je důkladně popsáno a tento příspěvek se jím nezabývá. Elektronovou hustotu v krystalu r(x,y,z) v místě daném frakčními souřadnicemi (x,y,z) počítáme z hodnot strukturních faktorů:
![]() , (3)
, (3)
kde sumace probíhá přes všechny indexy h, k, l a V je objem základní buňky. Strukturní faktor z předchozích dvou rovnic udává intenzitu záření rozptýlenou jednou základní buňkou krystalu a získá se jako součet atomových rozptylových faktorů, přičemž je nutno zauvažovat fázové rozdíly a teplotní pohyby atomů:
![]() . (4)
. (4)
Sčítá se přes všechny atomy j = 1, 2, ...N v buňce. fj je atomový rozptylový faktor udávající rozptyl záření na j-tém atomu, xj, yj a zj jsou frakční souřadnice atomu a Tj je teplotní faktor popisující vliv teplotního pohybu j-tého atomu. Atomový rozptylový faktor fj je spojen s elektronovou hustotou rj na j-tém atomu prostřednictvím Fourierovy transformace:
![]() , (5)
, (5)
kde S je difrakční vektor udávající směr difraktovaného záření a r je polohový vektor udávající polohu vzhledem k jádru j-tého atomu. Z rovnic (1-5) vyplývá, že nakonec je intenzita difraktovaného záření, měřená při difrakčních experimentech, funkcí elektronových hustot na jednotlivých atomech v krystalu. Různé modely popisované níže se liší právě tím, jakým způsobem definují funkci rj(r) popisující elektronovou hustotu na atomech:
|
Vztah
pro elektronovou hustotu na atomu |
Model |
Parametry |
Rovnice |
|
rj(r) = ratom(r) |
IAM |
xj,yj,zj,Tj |
(6) |
|
rj(r) = rcore(r) + rval(r,Pv,k) |
KR |
xj,yj,zj,Tj,Pv,k |
(7) |
|
rj(r) = rcore(r) + rval(r,Pv,k) + rdef(r,Plm±,k’) |
MR |
xj,yj,zj,Tj,Pv,k,Plm±,k’ |
(8) |
Tabulka 1
První model (rovnice 6) se nazývá model nezávislých atomů (Independent Atom Model, IAM) a je základem více než 99% rentgenostrukturních analýz. IAM předpokládá, že atomy v krystalu spolu nijak neinteragují, z čehož plyne, že jsou sféricky symetrické a nenabité. Tato aproximace se může zdát velice hrubá, ale funguje velmi dobře, neboť drtivá většina elektronové hustoty je skutečně sféricky symetrická a soustředěná v bezprostředním okolí jader atomů, jak je podrobněji diskutováno v ref. [1]. Parametry IAM modelu, které se upřesňují metodou nejmenších čtverců (Lest Squares Method, LS) v průběhu řešení struktury, jsou polohy atomů xj, yj, zj a teplotní parametry atomů Tj; soubor poloh atomů a teplotních parametrů je typickým výsledkem IAM (obr. 1).
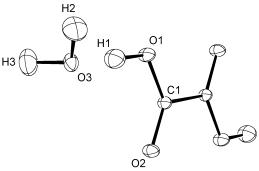
Obrázek
1. Struktura a-dihydrátu kyseliny oxalové [2]. Atomy se nacházejí s 50% uvnitř termálních
elipsoidů. Označeny jsou jen atomy ze symetricky nezávislé části, uprostřed C-C
vazby je krystalografický střed symetrie.
Druhý model (rovnice 7) je založen na kappa upřesňování (Kappa Refinement, KR, ref. [3]). KR předpokládá, že atomy v krystalu si mohou navzájem vyměňovat elektrony, takže mohou být kladně nebo záporně nabité a mohou též měnit svoji velikost. Atomy nicméně zůstávají sféricky symetrické, jak je patrné z pravé strany rovnice 7, která závisí pouze na vzdálenosti od jádra r, nikoli na vektoru r. V KR se rozdělí atomová el. hustota na elektronovou hustotu příslušející vnitřním a valenčním elektronům (rcore a rval). Dále se zavedou dva nové parametry: parametr Pv udává skutečný počet valenčních elektronů a parametr k popisuje rozšíření (k < 1) či smrštění (k > 1) valenční slupky vůči izolovanému atomu, pro který platí k = 1. Parametry modelu Pv a k se upřesňují naprosto standardním způsobem pomocí metody LS, společně s polohovými a teplotními parametry atomů. Z parametrů Pv je možno na konci KR vypočíst atomové náboje, které jsou typickým výsledkem této metody.
Třetí model (rovnice 8) je založen na tzv. multipólovém upřesňování (Multipole Refinement, MR, ref. [4]). MR dále rozšiřuje KR tak, že atomy si mohou nejen vyměňovat elektrony a měnit svoji velikost, ale mohou též měnit svůj tvar, tj. mohou jevit odchylky (deformace) od sférické symetrie, které jsou popsány prostřednictvím třetího členu rovnice 8. Parametry Plm± jsou tzv. multipólové populační parametry, které nabývají hodnot v intervalu <-1;1> a udávají jakým způsobem elektrony porušují sférickou symetrii, parametr k’ souvisí s roztažením či smrštěním deformací a má tedy pro třetí člen rovnice 8 analogický význam jako parametr k pro druhý člen. Všechny parametry modelu (tab. 2) se opět upřesňují metodou LS. Typickým výsledkem MR je v úplný popis elektronové hustoty v krystalu. Narozdíl od IAM a KR popisuje MR i tu část elektronové hustoty, která je v okolí atomů rozmístěna nesféricky, jako např. kovalentní vazby nebo volné elektronové páry (obr. 2). Odtud rovněž plyne, že funkce r(x,y,z) z IAM a KR jsou nekompletní a pro topologickou analýzu je vhodná jedině elektronová hustota z MR.
2.1.2. Praktické výpočty
Výpočty založené na IAM modelu reprezentují naprosto standardní rentgenostrukturní analýzu. K výpočtům je možno použít libovolný relevantní program, např. populární SHELX [5] nebo speciálnější JANA [6]. Funkce ratom(r) z rovnice 6, popisující elektronovou hustotu na izolovaných atomech jsou známy, jejich Fourierovou transformací dostaneme atomové rozptylové faktory fj (rovnice 5), které jsou vypočteny a tabelovány v mezinárodních krystalografických tabulkách [7] a příslušné programy [5,6], používají tyto funkce zcela automaticky, aniž by se o to uživatel musel starat.
K výpočtům pomocí modelů KR a MR již potřebujeme speciálnější software, který implementuje rozšíření naznačené v rovnicích 7 a 8. Lze použít původní program MOLLY [3], který je ovšem nepříliš uživatelsky přítulný, novější program XD [8], který je sice uživatelsky přítulnější, ale komerční a konečně program JANA [6], který je volně dostupný a patrně též nejrychlejší a uživatelsky nejpřítulnější. Dále bude popsáno jen MR, a to ze dvou důvodů: za prvé lze KR považovat za zjednodušenou variantu MR a za druhé KR poskytuje atomové náboje, ale ne úplný popis elektronové hustoty v krystalu, který je nutný pro TA.
Při MR upřesňování se postupuje zhruba následujícím způsobem:
(i) Naměříme difrakční data vhodná pro MR, tj. data dostatečně kvalitní (Robs maximálně kolem 4) vysokoúhlová (do sinq/l ³ 1.0 Å-1) a nízkoteplotní (nejčastěji se měří při teplotě kolem 100 K).
(ii) Vyřešíme strukturu standardním způsobem pomocí IAM modelu.
(iii) Změníme definice atomových elektronových hustot, tj. změníme fce ratom (rovnice 6) na funkce rcore a rval (rovnice 7 a 8).
(iv) Nadefinujeme lokální souřadné systémy (SS) pro jednotlivé atomy. K lokálním SS se vztahuje směr vektoru r z rovnice 8. Je též velmi vhodné nadefinovat lokální symetrii, která významně sníží počet parametrů Plm±. Například pro uhlíkový atom s hybridizací SP2 můžeme nadefinovat lokální trojčetnou osu. Více viz ref. [2].
(v) Upřesníme polohové a teplotní parametry atomů jen s použitím vysokoúhlových dat. Tento krok není nutný, ale je vhodný, protože při vyšších úhlech získáme polohové a teplotní parametry atomů méně ovlivněné vazebnými efekty, což může zlepšit stabilitu metody LS v následujících krocích (podrobněji viz [1,2,9]).
(vi) Zafixujeme polohy a teplotní parametry atomů a metodou LS postupně upřesňujeme další parametry z rovnice 8. Cílem postupného upřesňování je omezit korelace mezi parametry a tak zachovat stabilitu LS. Konečným cílem je upřesnit vše současně. V praxi se osvědčily různé modifikace následujícího postupu: upřesnit jen Pv, k, pak Pv, k, Plm±, pak jen Plm±, k’, pak vše současně (někdy s vynecháním parametrů k’). Jsou-li data extrémně kvalitní (což většinou nejsou), lze upřesňovat od začátku vše dohromady.
(vii) Specielní triky, přesahující rámec tohoto pojednání, vyžadují vodíkové atomy, u nichž je hlavní problém nepřítomnost vnitřních (core) elektronů (viz např. [2,3,10]).
(viii) Vykreslíme mapy elektronových hustot, které mohou být buď dynamické (= zahrnující teplotní pohyby atomů) nebo statické (= nezahrnující teplotní pohyby atomů).
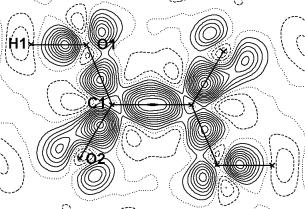
Obrázek 2. Dynamic
model deformation electron density map příslušející a-dihydrátu
kyseliny oxalové (srovnej s obr. 1).
Mapa je diferenční, takže ukazuje
rozdíl mezi celkovou elektronovou
hustotou a sférickou elektronovou hustotou na atomech. Výsledek rozdílu je nesférická
elektronová hustota na atomech, což jsou zde kovalentní vazby mezi atomy a
volné elektronové páry na atomech kyslíku O1 a O2. Vrstevnice na mapě jsou po
0.05 eÅ-3;
plné čáry reprezentují kladné, tečkované čáry nulové a přerušované čáry záporné
vrstevnice.
2.2. Nábojové hustoty a QM
2.2.1. Základní teorie
Elektronové hustoty v molekulách lze také spočítat pomocí metod kvantové mechaniky. Podle jednoho z postulátů QM je stav systému plně popsán jeho vlnovou funkcí Y. Vlnová funkce stacionárního systému, jako je například molekula v krystalu, může být vypočtena řešením časově nezávislé Schrödingerovy rovnice:
![]() , (9)
, (9)
která spojuje vlnovou funkci Y s kvantovanou energií systému E prostřednictvím Hamiltonova operátoru ![]() . Ačkoli je přesné řešení Schrödingerovy rovnice často
obtížné nebo dokonce nemožné, s použitím vhodných aproximací, moderních
programů a rychlých počítačů lze vypočítat dosti přesnou vlnovou funkci středně
velké molekuly během několika hodin. Jakmile je vypočtena vlnová funkce, lze z
ní odvodit další vlastnosti, jako například elektronovou hustotu, která je
úměrná čtverci absolutní hodnoty Y v daném
místě:
. Ačkoli je přesné řešení Schrödingerovy rovnice často
obtížné nebo dokonce nemožné, s použitím vhodných aproximací, moderních
programů a rychlých počítačů lze vypočítat dosti přesnou vlnovou funkci středně
velké molekuly během několika hodin. Jakmile je vypočtena vlnová funkce, lze z
ní odvodit další vlastnosti, jako například elektronovou hustotu, která je
úměrná čtverci absolutní hodnoty Y v daném
místě:
![]() . (10)
. (10)
2.2.2. Praktické výpočty
QM výpočty jsou dnes, díky neustálému zdokonalování hardwaru i softwaru, přístupné i ne-specialistům v oboru kvantové chemie. Ve skutečnosti jsou běžné QM výpočty z hlediska uživatele jednodušší než odpovídající XRD výpočty. QM výpočty jsou velmi vhodným doplňkem studií nábojových hustot, neboť umožňují ověření výsledků z nezávislého zdroje. Při porovnávání XRD a QM je vhodné si uvědomit důležitou skutečnost: XRD experimenty se týkají molekul v krystalech, zatímco běžné QM výpočty se týkají molekul ve vakuu. U molekulových krystalů, v nichž jsou molekuly vázány jen van der Waalsovými silami, můžeme tento rozdíl zanedbat a používat běžný QM software, jako je GAUSSIAN94 [11], který je v České republice dostupný pro akademické použití zdarma prostřednictvím virtuálního metapočítače [12]. Pro výpočet nábojů a elektronové hustoty v libovoné molekule stačí spustit v programu GAUSSIAN94 následující kód:
|
#SP |
|
#HF/6-31G** |
|
#P GFInput Iop(6/7=3) ... |
Obrázek
3. Výpočet elektronové hustoty v programu
GAUSSIAN94.
První řádek (obr. 3) udává typ výpočtu (SP = Single Point = výpočet jen pro jednu geometrii molekuly, žádné optimalizace), druhý řádek udává tzv. level of theory (level of theory je kombinace výpočetní metody (zde HF) a báze atomových orbitalů (zde 6-31G**)), třetí řádek obsahuje klíčová slova modifíkující výstup tak, aby bylo možno zobrazit funkci r(x,y,z) pomocí volně šiřitelného programu MOLDEN [13]. Zbytek souboru (schematicky naznačený třemi tečkami), obsahuje již jen řádek s libovolným titulkem, řádek s nábojem a multiplicitou a řádky s kartézskými souřadnicemi atomů.
3. Topologická analýza nábojových hustot
3.1. Základní pojmy
V kapitole 2 bylo naznačeno, jak získat elektronovou hustotu r(x,y,z), z XRD experimentu (rovnice 1-5, 8) nebo z QM výpočtu (rovnice 9-10). Jedná se o spojitou funkci tří proměnných. Zvolíme-li pro všechny tři proměnné krok 0.1 Å, pak dostaneme pro oxalovou kyselinu (obr. 1) přes 60 tisíc čísel, což je jistě báječné, ale uživatel si spíš klade otázky typu: který ze dvou atomů kyslíku nese větší záporný náboj? Je vazba mezi uhlíkovými atomy dvojná? Odpovědi na tyto otázky poskytuje topologická analýza [14].
Máme-li funkci r(x,y,z) = r(x1,x2,x3) = r(r), můžeme z ní vypočítat řadu dalších věcí: první derivace (dr(r)/dxi), druhé derivace (d2r(r)/dxidxj), gradient (Ñr(r)), laplacián (Ñ2r(r)), matici H(r) (matice 3x3, jejíž prvky jsou druhé derivace d2r(r)/dxidxj tzv. Hessian matrix), vlastní hodnoty H(r) (l1, l2, l3: tzv. křivosti (curvatures)). Zatím jsme jen zkomplikovali situaci dalšími horami čísel, ale v následujících krocích vše využijeme ke zjednodušení.
Gradient Ñr(r) je vektor, který ukazuje ve směru největšího nárůstu funkce r(r). Nyní začněme v libovolném místě r poblíž molekuly, vypočtěme gradient, posuňme se ve směru gradientu o element délky, vypočtěme další gradient, posuňme se atd. Takto nakonec dostaneme křivku, která dříve nebo později skončí na nějakém atomovém jádře, kde je lokální maximum funkce r(r) a odkud už není kam jít. Všechny body r z okolí molekuly, ze kterých se tímto způsobem dostaneme k danému jádru, jednoznačně vymezují jakousi oblast (basin) příslušející danému atomu. V této oblasti provedeme integraci elektronové hustoty a dostaneme atomový náboj.
Ukazuje se, že nejdůležitějšími prvky funkce r(r) jsou takzvané kritické body (Critical Points; CP), kterých je v každé molekule či elementární buňce jen několik a nacházejí se v místech, kde jsou všechny první derivace nulové (dr(r)/dxi = 0). CP se dále charakterizují pomocí pomocí křivostí l1, l2, l3: každý CP je označen dvěma čísly, z nichž první udává počet nenulových křivostí (v případě stabilní molekuly vždy 3) a druhé udává součet znamének křivostí (zde jsou čtyři možnosti, viz např. [14,15]). Každé mapě elektronové hustoty dominují maxima na atomových jádrech, což jsou podle TA kritické body (3,-3). Z chemického hlediska jsou nejzajímavější sedlové body vazeb, z hlediska TA jde o CP (3,-1), tzv. kritické body vazby (Bond Critical Points; BCP). Pomocí hodnot výše definovaných funkcí v místě BCP lze každou vazbu kvantitativně popsat. Např. dvojný charakter vazby lze získat pomocí hodnoty veličiny zvané elipticita (elipticity: e = l1/l2 - 1) v BCP, která se liší od nuly tím víc, čím víc se vazba blíží dvojné.
3.2. Praktické výpočty v XRD
Máme-li vypěstován nadprůměrně kvalitní krystal (což je patrně nejobtížnější), změřena vysoce kvalitní difrakční data (rovněž netriviální úkol) a provedené multipólové upřesnění (relativně snadné, viz kapitola 2.1.2) jsou výpočty týkající se topologické analýzy v programu JANA z uživatelského hlediska velmi jednoduché. Pro výpočet atomových nábojů stačí zadat z grafického menu: Tools ® Topological analysis ® Basin integration a projít několika intuitivními dialogovými okny. Analogicky pro vyhledání CP a výpočet všech příslušných hodnot stačí v JANĚ zadat: Tools ® Topological analysis ® Find critical points.
3.3. Praktické výpočty v QM
Výpočet atomových nábojů přímou integrací funkce r(x,y,z) a všech patřičných veličin ve všech CP libovolné molekuly zajistí v programu GAUSSIAN94 následující kód:
|
#SP |
|
#HF/6-31G** |
|
#AIM=(Charges,CP) ... |
Obrázek
4. Výpočet atomových nábojů a CP v programu
GAUSSIAN94.
První dva příkazy na obr. 4 jsou identické s obr. 3 (kapitola 2.2.2), příkaz na třetím řádku říká programu, že má provést topologickou analýzu s výpočtem nábojů a určením kritických bodů, zbytek souboru na obr. 4 je opět totožný s obr. 3.
4. Závěr
Tento příspěvek, vzhledem ke své omezené délce, nemůže pokrýt celou problematiku spojenou s nábojovými hustotami a topologickou analýzou a suplovat tak objemné učebnice [15, 16] či obsáhlé souhrné články [17, 18]. Jedná se o velmi stručný úvod do problematiky, která může být zajímavá esteticky (viz obr. 2), teoreticky (přímé srovnání QM a XRD) i prakticky (využití informací o atomových nábojích při syntézách). Vše bylo velmi zjednodušeno, úplně vynechána byla mj. pasáž týkající se X-X a X-N upřesňování [9] a klíčová pasáž zabývající se měřením XRD dat vhodných pro studium nábojových hustot (viz minulé kolokvium, ref. [19]). Topologická analýza [14] byla jen naznačena. Pro podrobnější informace je třeba odkázat na výše zmíněné učebnice a články [15-18], popřípadě i na autorovu disertační práci [2] a souhrnné články v češtině [19, 20].
Literatura
[1] V. Valvoda: Experimentální metody studia pevných látek. Rentgenová difrakce: struktury, nábojové hustoty, vazby. (Lukáč P., ed.), sv. 3, Matematicko-fyzikální fakulta UK, Praha 1981.
[2] M. Šlouf: Disertační práce & odkazy v ní uvedené. Karlova univerzita, Praha 2001.
[3] P. Coppens, T.N. Guru Row, P. Leung, E.D. Stevens, P.J. Becker & Y.W. Yang, Acta Cryst. A35,(1979) 63.
[4] N.K. Hansen & P. Coppens, Acta Cryst. A34 (1978) 909.
[5] G.M. Sheldrick (1997). SHELX-97. Programs for X-ray structure determination and refinement. University of Gottingen, Germany.
[6] V. Petříček & M. Dušek (2000). JANA2000. Crystallographic Computing System. Institute of Physics, Praha, Czech Republic.
[7] International Tables for Crystallography (1995). Vol. C, edited by A.J.C. Wilson. Dodrecht: Kluwer.
[8] T. Koritsanszky, S. Howard, Z. Su, P.R. Mallinson, T. Richter, & N.K. Hansen (1997). XD. Computer Program Package for Multipole Refinement and Analysis of Electron Densities from Diffraction Data. Free University of Berlin, Germany.
[9] J. Loub, Chem. Listy 78 (1984) 720.
[10] M. Šlouf, A. Holý, V. Petříček, & I. Císařová, Acta Cryst. B58 (2002) 519.
[11] M.J. Frisch, G.W. Trucks, H.B. Schlegel, P.M.W. Gill, B.G. Johnson, M.A. Robb, J.R. Cheeseman, T. Keith, G.A. Petersson, J.A. Montgomery, K. Raghavachari, M.A. Al-Laham, V.G. Zakrzewski, J.V. Ortiz, J.B. Foresman, J. Cioslowski, B.B. Stefanov, A. Nanayakkara, M. Challacombe, C.Y. Peng, P.Y. Ayala, W. Chen, M.W. Wong, J.L. Andres, E.S. Replogle, R. Gomperts, R.L. Martin, D.J. Fox, J.S. Binkley, D.J. Defrees, J. Baker, J.P. Stewart, M. Head-Gordon, C. Gonzalez, and J.A. Pople, Gaussian 94, Revision D.4, Gaussian, Inc., Pittsburgh PA, 1995.
[12] http://meta.cuni.cz/
[13] G. Schaftenaar & J.H. Noordik, J. Comput.-Aided Mol. Design, 14 (2000) 123.
[14] R.F.W. Bader: Atoms in Molecules: a Quantum Theory. Oxford 1990. Clarendon Press, Oxford Science Publications.
[15] P. Coppens: X-ray Charge Densities and Chemical Bonding. New York 1997. Oxford University Press.
[16] V.G. Tsirelson & R.P. Ozerov: Electron Density and Bonding in Crystals. Bristol, England / Philadelphia, PA 1996. Institute of Physics Publishing.
[17] P. Coppens, Acta Cryst. A54 (1998) 779.
[18] G.U. Kulkarni, R.S. Gopalan & C.N.R. Rao, J. Mol. Struct. (Theochem) 500 (2000) 339.
[19] M. Šlouf, Materials Structure, 9(1) (2002) 26.
[20] M. Šlouf, Chem. Listy 96 (2002) 3.