ŘEŠENÍ KRYSTALOVÝCH STRUKTUR
Z PRÁŠKOVÉ DIFRAKCE
Radovan Černý
Laboratoire de
Cristallographie, Université de Genève, 24 quai Ernest-Ansermet, CH-1211 Genève
4, Suisse
Úvod
Ne vždy máme pro určení
neznámé krystalové struktury k dispozici kvalitní monokrystal. Typickým
případem jsou kovové slitiny a jejich intermetalické fáze, hydridy techto
fází, některé molekulární krystaly a řada dalších. Byly to
molekulární materiály, zvláště farmaceutické látky, které motivovaly
v posledních 20 letech intenzivní vývoj v teorii i experimentu
difrakčních metod používajících polykrystalický vzorek. Celkový počet
krystalových struktur určených těmito metodami se pohybuje někde
okolo 600 s roční produkcí vyšší než 50 (http://www.cristal.org/iniref.html).
A přiznejme si, že vzrůstající síla těchto metod někdy vede
k poklesu intensity práce v přípravě materiálu, neboť
je rychlejší připravit polykrystalický vzorek než strávit řadu
týdnů na přípravě dostatečně velkého monokrystalu (i
když monokrystalové difraktometry při zdrojích synchrotronového
záření posouvají potřebnou velikost monokrystalu stále dolů).
Prášková difrakce nemůže nikdy dosáhnout prostorového rozlišení a strukturní
přesnosti difrakce monokrystalové. Pracuje totiž s 1-rozměrnou
projekcí původně 3-rozměrného difrakčního obrazu
v reciprokém prostoru, která nutně vede k překryvu
nezávislých informací i v ideálním případě difraktometru s nekonečně
velkým úhlovým rozlišením. Prášková difrakce se může ve výkonosti pouze približovat
difrakci monokrystalové a může těžit ze širokého arzenálu metod zde
již existujících. Nižší rozlišení měřených dat práškové difrakce však
vedlo k vývoji netradičních metod řešení struktur (globální
optimalizace), které našly svou inspiraci v jiných oblastech výzkumu setkávajících
se s podobným problémem: Obtížností přímé cesty od dat, která je
možno měřit, k hodnotám hledaných parametrů.
|
|
|
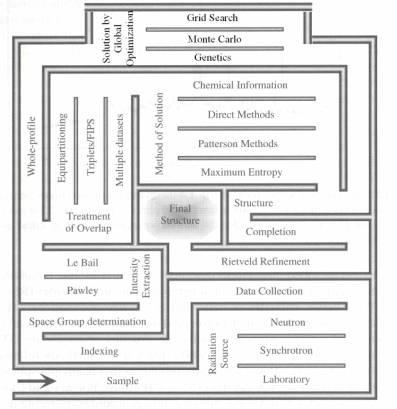
Obr. 1: Bludiště
řešení krystalové struktury; modifikováno z [1]. |
Současný limit práškové difrakce je asi 30
atomů v asymetrické buňce pro rutinní práci, struktury se 60
atomy už byly vyřešeny. Optimisté odhadují, že brzy můžeme dosáhnout
limitu 100 atomů (http://www.sacrs.org.za/ecm21/plenary.html).
Řešení krystalové struktury může být přirovnáno k bludišti
(obr. 1): I když k cíli (struktuře) vede více cest, ne všechny jsou
vhodné či dokonce možné pro daný problém. Základními etapami, komorami
v bludišti, však musí projít každý, má však možnost volby mezi
různými cestičkami. O některých z nich pojednává tento
příspěvek. Nebudeme se zabývat přípravou vzorku a optimalizaci
difrakčního experimentu ani poslední etapou – vypřesnění
struktury pomocí Rietveldovy metody.
Indexace práškového
difrakčního záznamu
První, a ne vždy lehký krok, je
určení elementární buňky našeho neznámého krystalu – indexace
difrakčního záznamu. Ve srovnání se monokrystalovými daty je to úloha o to
těžší, že nemáme k dizpozici informaci o orientaci reciprokých
vektorů r*hkl, ale jenom o jejich délce 1/dhkl.
Základní soustavou rovnic, se kterou pracují všechny indexační algoritmy
práškových dat je
Qhkl
= 1/d2hkl = h2a*2 + k2b*2
+ l2c*2 + 2hka*b*cosg* + 2klb*c*cosa* + 2hla*c*cosb* (1)
čili pro každou pozorovanou hodnotu Qhkl
(danou polohou jedné reflexe hkl v práškovém difrakčním
záznamu) máme najít tři celá čísla (Millerovy indexy hkl) a
šest mřížových parametrů stejných pro celou soustavu rovnic. Rovnice
(1) nabývá jednodušších forem pro vyšší symetrie krystalové mříže. Hodnoty
Qhkl jsou měřeny s určitou přesností,
tudíž teoreticky by bylo možné oindexovat každý difrakční záznam pomocí
kubické elementární buňky s mřížovým parametrem ~ 105 Å, což samozřejmě není
správné řešení. Čili bez ohledu na to jak indexujeme difrakční
záznam, potřebujeme jednoduché kriterium pro fyzikální správnost řešení.
Takový parametrem je de Wolffovo kriterium (Figure-of-Merit)
M20 = Q20
/ (2 <Q> N20) (2)
kde pracujeme s prvními 20 pozorovanými a
oindexovanými reflexemi, Q20 je Q hodnota pro 20-tou
reflexi, <Q> je průměrný rozdíl
mezi pozorovanými a spočtenými Q hodnotami a N20
je počet spočtených reflexí až do pozorované 20-té reflexe. Hodnota M20
> 10 značí s velkou
pravděpodobností fyzikálně správné řešení. Jiným kriteriem je
Smithův a Snyderův F-index
FN
= 1 / <D2q> × N / N(qg) (3)
kde pracujeme s pozorovanými reflexemi až do
úhlu qg, N je počet
pozorovaných a N(qg) počet
spočtených reflexí až do tohoto limitu a <D2q> je průměrný rozdíl
mezi pozorovanými a spočtenými hodnotami 2q. Na rozdíl od M20 nezávisí FN
na symetrii krystalové mříže, což však není vždy výhoda neboť kubické
řešení je pravděpodobnější než triklinické pro stejný počet
oindexovaných reflexí a stejnou přesnost poloh reflexí. Pro spravný
výpočet M20 a FN by měly být
vzaty v úvahu systematické absence, což je možné až po určení
prostorové grupy. Úspěšnost indexace, tak jako celého procese určení
struktury, závisí silně na kvalitě měřených dat
včetně včasné identifikace reflexí příměsových fází.
Obtížný je případ indexace krystalové mříže s dominantní zonou –
jedna krystalová osa podstaně kratší než ostatní dvě.
Vlastní indexační proces
používá různé algoritmy jak vyřešit soustavu, typicky 20-ti, rovnic
(1) a provádí inteligentní nebo vyčerpávající hledání v přímém (mřížové
parametry) nebo reciprokém (Millerovy indexy) prostoru. Program ITO [2] využívá
toho, že reflexe náležijící stejné centrální zoně (rovina
v reciprokém prostoru procházející počátkem) lze oindexovat pomocí
pouze dvou indexů a hledá takové zony v pozorovaných datech. Program
DICVOL91 [3] provádí vyčerpávající hledání v přímém prosotoru a
využívá přitom principu dichotomie: systematicky hledá oblasti hodnot
mřížových parametrů, které mohou obsahovat řešení a
vylučuje ostatní oblasti. Program TREOR90 [4] provádí vyčerpávající
hledání v reciprokém prostoru a využívá přitom určitého
stupně inteligence plynoucího z předchozích zkušeností. CRYSFIRE (http://www.ccp14.ac.uk/tutorial/crys/index.html
) je rozhraní k více indexačním programům s analyzou
výsledků. Jelikož řešení mnohaparametrové soustavy rovnic (1) je
typický problém globální optimalizace, není překvapující, že se objevila
řada indexačních programů uživajících algoritmy globální optimalizace
jako metody Monte Carlo, genetický algoritmus a jiné. Přehled všech metod a
odkazy na jednotlivé programy lze najít v (http://www.ccp14.ac.uk/solution/indexing/index.html).
Vlastní proces úspěšné
indexace musí být zakončen prověřením správnosti nalezené
elementární buňky pomocí Whole-Pattern-Profile-Fitting, tak jak je popsán
v následující kapitole, a rovněž i redukcí buňky a nalezením
buňky Bravaisovy, která vykazuje přijatelné systematické absence.
Určení prostorové grupy a
integrovaných intenzit jednotlivých reflexí
Základní problem práškové
difrakce, 1-rozměrná projekce původně 3-rozměrného
difrakčního obrazu, vede k částečnému či úplnému
překryvu reflexí v difrakčním záznamu. Obtížnost pracovat
s integralními intenzitami jako s nezávislými pozorováními vedla H.
Rietvelda [5] ke slavné metodě nesoucí dnes jeho jméno, modelování celého
digitálního práškového difrakčního záznamu, a užití každého bodu jako
nezávislého pozorování. Tento přístup lze také nazvat
Whole-Pattern-Profile-Fitting (WPPF) a má dvě varianty: (a) užívající
strukturní model – vlastní Rietveldova metoda, (b) bez strukturního modelu –
metoda Le Bailova [6] nebo Pawleyho [7]. Je dobré si uvědomit, že (a) je
ve své podstatě vypřesňovnání strukturních parametrů
z dat práškové difrakce, zatímco (b) slouží hlavně k dekompozici
práškového difrakčního záznamu na jednotlivé integrální intenzity.
Obě metody, (a) i (b), však mají mnoho společného, takže je vždy
najdeme jako součast jednoho výpočetního programu. Nebudeme zde
podávat matematický základ Rietveldovy metody, to lze najít
v příslušné kapitole tohoto přehledu, budeme se zde zabývat
pouze metodou (b) neboť lze pomocí ní ověřit správnost indexace,
získat informace o systematických absencích – prostorové grupě krystalu a
získat jednotlivé integrální intenzity.
Metoda Le Bailova je WPPF bez
strukturního modelu, avšak s mřížovými parametry a prostorovou grupou
(pro začátek to může být grupa holoedrie). Mřížové parametry,
profilové parametry (šířky a tvar reflexí) a instrumentální parametry
(kalibrace 2q, pozadí) jsou upřesňovány
(např. metodou nejmenších čtverců) a hodnoty integrálních
intenzit jsou upřesňovány pomocí iterativní metody založené na
původním Rietveldově vztahu pro výpočet pozorovaných
integrálních intenzit. Při iterování se pozorované intenzity z n-té
iterace stávají vypočtenými intenzitami pro (n+1)-tou iteraci,
přičemž počáteční hodnoty vypočtených intenzit
v 0-té iteraci odpovídají např. rovnoměrnému rozdělení
celkové intenzity mezi překrývající se reflexe. Metoda Pawleyho se liší
tím, že integrální intenzity jsou považovány za volné parametry a jsou
upřesňovány současně se všemi ostatními volnými parametry.
Úspěšně provedené La
Bailovo nebo Pawleyho vypřesnění slouží v prvé řadě
pro prověření správnosti elementární buňky, dále pro získání
informací o systematických absencích a nakonec i pro získání hodnot
pozorovaných integrálních intenzit Ihkl(obs)
jednotlivých reflexí pro jejich další použití při určení krystalové
struktury pomocí metod monokrystalových. Určení prostorové grupy
z práškových dat se ve své podstatě řídí stejnými pravidly jako
pro data monokrystalová. Překryv jednotlivých reflexí v práškovém
difrakčním záznamu však způsobí omezení pouze na nízkoúhlové reflexe
a prakticky znemožní určení ze statistiky intenzit zda prostorová grupa
obsahuje střed symetrie. Užití celého difrakčního záznamu a zkoumání
systematických absencí pomocí Bayesovy teorie pravděpodobnosti [8] znamená
mnohem kompletnější analyzu. Pro určení integrálních intenzit Ihkl(obs)
pomocí WPPF musíme rozhodnout jak budeme zacházet
s částečně a úplně se překrývajícími reflexemi. O
nových metodách založených na užití dodatečné krystalografické informaci
anebo na dodatečném experimentu pojednává následující kapitola.
Metody určení integrovaných
intenzit překrývajících se reflexí
Užití dodatečné
krystalografické informace (atomicita a pozitivnost)
Během procesu určovaní
fází reflexí jejichž integrální intenzity již byly spolehlivě určeny
(nepřekrývající se reflexe) získáme řadu dodatečných informací,
které můžeme použít pro separaci překrývajících se reflexí. Informace
o pseudo-translační symetrii byla použita v programu EXPO [9]: Překrývají-li
se reflexe super-struktury s reflexí sub-struktury, je
pravděpodobnější, že druhá bude mít větší intenzitu. Užití
fázové informace již lokalizované částečné struktury (molekulární
fragment) se provádí ve stejném programu. Uzití informace o strukturních
invariantech se užívá v programu DOREES-POWSIM [10]: Známe-li již moduly
dvou reflexí v silném tripletu çEkça çEh+kçmůžeme s velkou pravděpodobností
tvrdit, že modul třetí reflexe v tripletu, çEhç, bude rovněž velký.
Očekávaná pozitivnost
Pattersonovy funkce v přímém prostoru: Pattersonova funkce spočtena
z dat, která jsou k dispozici (nepřekrývající se reflexe), je
ne-lineárním způsobem
modifikována, přičemž je Pattersonově funkci vnucena
pozitivnost. Z této modifikované Pattersonovy funkce pak můžeme
zpětnou transformací obdržet druhé mocniny modulů všech reflexí, tedy
i překrývajících se. Tyto nové odhady pak mohou sloužit jako nové
počáteční hodnoty pro Le Bailovu metodu anebo celý proces může
být iterativně opakován. Pattersonova mapa může být modifikována na
základě principu maximální entropie [11] anebo na základě Sayerova
argumentu „Druhá mocnina Fourierovy mapy je podobná Fourierově mapě
samotné“ aplikovaného v iterativní podobě jako Fast-Iterative-Patterson-Squaring
v [12].
Užití dodatečného
experimentu – více difrakčních záznamů
Měřením více
difrakčních záznamů na jednom vzorku při různých, ale
kontrolovaných, vnějších podmínkách můžeme získat více informace o
intenzitách překrývajících se reflexí. Vztah mezi difrakčními záznamy
musí být znám anebo měřitelný. Dva případy měněných
vnějších podmínek byly úspěšně aplikovány: teplota a textura.
Máme-li materiál
s anisotropní teplotní roztažností potom relativní intenzity reflexí se
téměř s teplotou nemění, avšak jejich relativní polohy ano.
Tím se mění i obraz překryvu reflexí a měřením difrakčních
záznamů při různých teplotách obdržíme dodatečnou
informaci, která může pomoci separaci reflexí. Tato myšlenka byla poprve
použita již v [13] a v moderní podobě v [14].
Tak jako anisotropní teplotní
roztažnost mění anisotropně polohy reflexí s teplotou, tak textura
(přednostní orientace krystalitů) mění anisotropně
intenzity reflexí se změnou orientace difrakčního vektoru. Podmínkou
využití textury k separaci překrývajících se reflexí je texturovaný
vzorek a znalost textury (např. jejím změřením na nepřekrývajících
se reflexích). Máme-li potom n překrývajících se reflexí tak, že
můžeme pouze určit jejich souhrnou intenzitu Itot, můžeme
získat jejich integrální intenzity Ii (i = 1 ... n)
řešením soustavy rovnic (4) můžeme-li určit texturní korekci Ti(aj). Parametr aj označuje m měření
difrakčního záznamu při m různých orientacích
difrakčního vektoru vůči vzorku, např. náklon vzorku o úhel
aj na texturním goniometru.
Itot (a1) = I1 T1(a1) + I2 T2(a1) + ...
+ In Tn(a1)
Itot
(a2) = I1 T1(a2) + I2 T2(a2) + ...
+ In Tn(a2)
.............................................................................................. m ≥ n (4)
Itot (am) = I1 T1(am) + I2 T2(am) +...
+ In Tn(am)
Textura jako nástroj pro zisk
maximální informace z práškové difrakce byla navržena a poprvé využita v
[15] a úspěšně použita v [16, 17].
Metody řešení krystalové
struktury vyžadující integrované intenzity – metody reciprokého prostoru
Přímé metody
Přímé metody založené na
teorii pravděpodobnosti fází pozorovaných strukturních faktorů
známe-li jejich moduly, se vyvinuly v mocný nástroj určování
krystalových struktur z monokrystalových dat. Nic nám nebrání, pokud se
nám podaří získat integrální intenzity jednotlivých reflexí, aplikovat
stejné metody na prášková data. Přímé metody vyžadují pro svou
úspěšnou funkci kvalitní data s rozlišením odpovídajícím typické
meziatomové vzdálenosti v krystalu (~1 Å pro organické látky, nižší
rozlišení pro latky s těžkým atomem), což může být problém pro
prášková data díky vzrůstajícímu překryvu reflexí se vrůstajícím
difrakčním úhlem. Díky menší přesnosti modulů strukturních
faktorů získaných z práškových dat a jejich možnému systematickému
ovlivnění metodou separace překrývajích se reflexí, standardní
statistické testy přímých metod (Wilsonův test, rozdělení
normalizovaných modulů, Figures-of-Merit) nemusí vždy spolehlivě
fungovat. Dva výpočetní programy přímých metod byly optimalizovány
pro užití práškových dat, POWSIM [10] a EXPO [9]. Dekompozice práškového záznamu
je jejich integrální součástí, tak jak je diskutováno
v předchozí kapitole. Jiný způsob využití teorie, která je
základem přímých metod, je maximalizace Sayreovy rovnice vzhledem
k fázím, kde je v součtové funkci modulů Pattersonova typu
odstraněno maximum v počátku [18].
Pattersonova funkce
Pattersonova funkce byla
s úspěchem použita již v raném období práškové difrakce, protože
zvláště při aplikaci na struktury obsahující těžký atom je tato
metoda robustní a funguje i s daty s nízkým rozlišením. Čitelnost
Pattersonových map je však omezena jejich “rozmazáním“ díky problemům
uvedeným v předchozím odstavci. Čitelnější mapy lze obdržet
z neutronové difrakce zvláště v případě
přítomnosti atomu s negativním rozptylovým faktorem. Elegantní
způsob “zaostření“ Pattersonových map z práškové difrakce je
uveden v [11]. Celková intenzita skupiny překrývajících se reflexí je
považována za jeden údaj a digitalizována Pattersonova mapa je rekonstruována
na principu maximální entropie tak jak je známo v metodách rekonstrukce
obrazu. Stejná procedura může rovněž sloužit ke zlepšení
rozdělení celkové intenzity skupiny mezi jednotlivé reflexe, tak jak je
uvedeno v předchozí kapitole.
Maximalizování entropie
Princip maximalizování entropie
může být uplatněn přímo v procesu konstrukce map
elektronových hustot. Ukázalo se [19], že tento přístup je obzvláště
užitečný pro data z nižším rozlišením - data z práškové
difrakce. Fázování všech strukturních faktorů ze základního souboru se
provádí na základě maximalizování vhodné pravděpodobnostní
(likelihood) funkce a stejný princip lze použít i pro separaci
překrávajících se reflexí. Celý proces lze najít ve výpočetním
programu MICE [20].
Metody řešení krystalové
struktury globální optimalizací strukturního modelu a kompletního
difrakčního obrazu – metody přímého prostoru
Základní problém práškové
difrakce, obtížnost získání kvalitních odhadů intenzit jednotlivých
reflexí, lze obejít tím, že se o žádné odhady nebudeme pokoušet. Místo analýzy
(rozkladu) difrakčního obrazu přejdeme k jeho syntéze (simulaci)
s “náhodným“ (zde můžeme vložit do procesu dodatečnou chemickou
či jinou informaci a zvýšit tak jinak značně deficitní
informační obsah dat) strukturním modelem, který dále optimalizujeme
vhodným algoritmem globální optimalizace. Můžeme na základě
Rietveldova principu porovnávat celý difrakční profil, anebo počítat
jenom integrované intenzity a porovnávat je s korelovanámi integrálními
intenzitami získanými z WPPF. Pro rozhodování o vhodnosti (fitness)
aktuálního strukturního modelu musíme zavést funkci, která bude vhodnost charakterizovat
- cost function (CF). Může to být např. parametr Rwp
známý z Rietveldovy metody nebo lépe parametr c2
c2 = D/(M - N) D = åi=1…M wi [yobsi
- ycali]2 (5)
kde yobs a ycal
jsou pozorované, s vahou w, a spočtené intenzity v bodě i
digitálního difrakčního záznamu a D je kvadratická forma minimalizovaná
v Rietveldově metodě pomocí metody nejmenších čtverců
pro M pozorování a N volných parametrů. Parametr c2 je vhodnější
z důvodu správnější vážené kombinace CF založené na
difrakčních datech s CF založené na dodatečných informacích.
Při práci s korelovanými integrálnímí intenzitami je takovou CF
např. [21]
c2 = åh åk [(Iobsh -
Icalh) (Vhk)-1 (Iobsk
- Icalk)] (6)
kde Iobsh a Icalh
jsou pozorované a spočtené integrální intenzity reflexe h a Vhk
je matice kovariancí integrálních intenzit reflexí h a k
z WPPF (Pawleyho metoda).
CF založené na difrakčních
datech mohou být kombinovány s CF založenými na dodatečných
pozorováních či chemických informacích. Kombinování (např. součet
CF) musí být váženo, správná kvantifikace váh je stále objektem diskuse
v literatuře. Nejčastěji používanou ne-difrakční CF je
energie krystalové mříže, nejčastěji pro tyto účely
popisovaná potenciální energií aktuálního uspořádání atomů. Potenciální
energie může být modelována nejjednoduššeji Coulombovskou interakcí (látky
z iontovou vazbou) nebo popsána vhodným potenciálem (Buckingham, Leonard-Jones
nebo Born-Mayer) za předpokladu, že vhodné parametry potenciálu jsou známé
pro daný problém [22]. Pro většinu intermetalických látek stačí popis
meziatomové interakce pouze pomocí její repulsivní části – minimální
meziatomové vzdálenosti (anti-bump); každý strukturní model obsahující
příliš krátké meziatomové vzdálenosti je penalizován vysokou hodnotou této
CF. Výpočty ab-initio krystalové energie jsou ve většině
případů příliš časově náročné pro jejich použití
v globální optimalizaci. Energie krystalové mříže jako součást
CF je používaná ve výpočetních programech [23-26], anti-bump CF jsou pak k dizpozici téměř ve všech
programech.
Aktuální strukturní model se
skládá z jednotlivých strukturních bloků: izolovaných atomů,
molekulárních fragmentů, celých molekul, koordinačních
mnohostěnů aj. Je výhodné použít jednotný popis strukturních
bloků pomocí vnitřních souřadnic (stereochemical descriptors)
jako vazebné vzdalenosti, vazebné úhly a torzní úhly. Polohu a orientaci celého
bloku pak pomoci tří krystalografických souřadnic (popisujících
polohu těžiště anebo jednoduššeji polohu vybraného atomu) a tří
úhlů (např. Eulerových). Výhody jsou hned dvojí: nižší počet
volných parametrů N (pro popis koordinačního
čtyřstěnu potřebujeme 5´3=15 krystalografických
souřadnic jednotlivých atomů, ale jenom 3+3=6 polohových a
orientačních souřadnic bloku) a jejich přímý
fyzikálně-chemický význam umožňující vložení jejich správných hodnot
do strukturního modelu. Hodnoty vnitřních souřadnic mohou být
buď pevné (rigid body) anebo proměnné v předem odhadnutelném
(např. ze znalosti struktur příbuzných krystalů) a většinou
úzkém (ne však pro torzní úhly v molekulách) intervalu. Vnitřní
souřadnice bloků mohou být obdrženy z kartezských souřadnic
daného bloku [27] nebo uloženy v tzv. Z-maticích (http://chemistry.umeche.maine.edu/Modeling/GGZmat.html).
Algoritmus globální
optimalizace
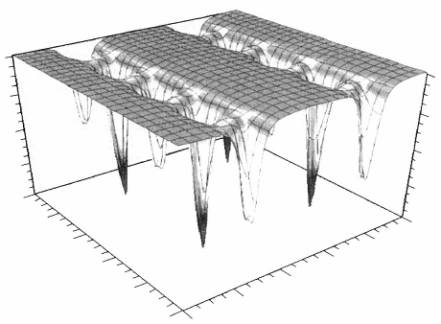
Úkolem algoritmu je
najít globální minimum celkové CF, kterou si můžeme představit jako
hyperplochu v N-rozměrném prostoru (N parametrů,
které určujeme). Příklad dvojrozměrného řezu takovou
hyperplochou je znázorněn na obr. 2.
Obr. 2: Dvojrozměrný
řez 12-rozměrnou hyperplochou c2 famotidinu B ;
převzato z [1].
Jak je vidět, hyperplocha CF
obsahuje řadu lokálních minim a algoritmus musí být schopen najít minimum
globální (odpovídající spravnému strukturnímu modelu), ale i být schopen
uniknout z minima lokálního. Je vidět, že jakýkoli algoritmus
kontrolovaný pouze gradientem hyperplochy (např. metoda nejmenčích
čtverců) skončí dříve či později v lokálním
minimu. Je to typický problém globální optimalizace a krystalografie zde našla
řadu inspiraci v ostatních vědách jako teoretická fyzika,
sociologie (modelování složitých společenských systemů) či
ekonomie (předpovídání ekonomických vývojů). Pro více informací o
globální optimalizaci lze doporučit [28]. Je nutné zdůraznit, že
každý algoritmus globální optimalizace je výpočetně nesmírně
náročný. První příklady řešení struktur globální optimalizace
strukturního modelu jsou spojeny s předpovídáním existence nových
látek na základě minimalizace krystalové energie a prášková difrakční
data byla užívána spíše pro kontrolu správnosti modelu a posteriori [29,
30].
1. Trial-and-error, grid
search
První práce řešení struktur
globální optimalizací s difrakčními daty užívaly jednoduše metodu zkoušky
a omylu (trial-and-error) [31], která se, zjednodušeně řečeno,
vyvinula v systematické prohledávání objemu elementární buňky [32, 33].
Oba přístupy však brzy narazily na hranice svých možností a bylo jasné, že
je musí nahradit “inteligentnější“ algoritmy.
2. Monte Carlo -
simulované žíhání
Termín “Monte Carlo“ je
v literatuře rezervován pro metodu vzorkování určitého prostoru
parametrů (hyperplochy) pomocí náhodných kroků zajištujících ergodicitu
vzorkování, t.j. každý bod hyperplochy může být dosažen se stejnou
pravděpodobností. Je to globální vzorkování nikoliv optimalizace. Teprve
při užití určité “inteligence“, jako např. simulované žíhání se
algoritmus stává globální optimalizací. Simulované žíhání se
nejčastějí provádí pomocí Metropolisova algoritmu [34] navrženého
při simulaci chování soustavy pevných koulí - plynu. Schema algoritmu je
na obr. 3.
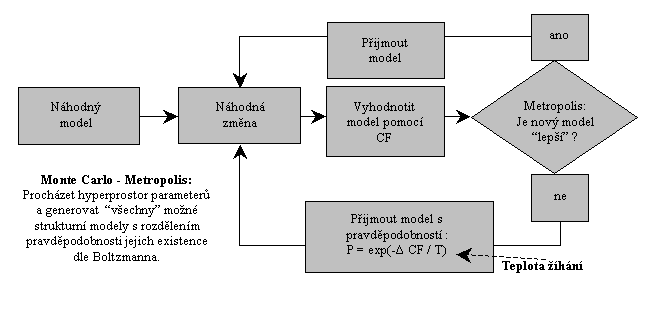

Obr. 3: Schema Monte-Carlo optimalizace s Metropolisovým algoritmem.
Pravděpodobnost
přijmutí “horšího“ modelu stoupá se teplotou žíhání T a
zajišťuje, že algoritmus nezůstane během svého optimalizovaného
vzorkování uvězněn v lokálním minimu hyperplochy CF. Optimalizace
začíná s vysokou teplotou T a s ní i spojenými velkými
náhodnými změnami modelu (změny N volných parametrů). To
zajišťuje rychlé avšak hrubé vzorkování hyperplochy. V dalších
krocích algoritmu se teplota postupně snižuje s cílem jemněji
vzorkovat hyperplochu v okolí již globálního minima, kam algoritmus
mělo zavést žíhání při vyšší teplotě. Odtud název algoritmu. První
užití Monte Carlo optimalizace pro řešení struktur z práškových difrakčních
dat [35] užívalo žíhání při konstantní teplotě T. Simulované
žíhání s různými způsoby kontroly postupného snižování teploty T
je v současné době nejrozšířenější metoda globální
optimalizace aplikovaná na řešení struktur z práškových dat [23-25, 36-41].
Při aplikování simulovaného žíhání je nutno správně odhadnout
rychlost “chlazení“, která závisí na počtu volných parametrů N
a také na kvalitě dat. Velká rychlost vede k “zakalení“ vysokoteplotního
modelu (systém zůstane v lokálním minimu), nízká rychlost vede ke
neúměrně dlouhým časům optimalizace (je nutno říci, že
ne každý běh algoritmu vede do globálního minima; je to přece jenom
algoritmus založený na náhodnosti). Proto byla v [42] navržena alternativa
algoritmu známá jako paralelní žíhání, které probíhá souběžně
při několika teplotách Ti rozdělěných
mezi nejnižší a nejvyšší vhodnou teplotu a algoritmus pravidelně porovnává
nejlepší řešení (strukturní modely) v sousedních (vzhledem k
teplotě) optimalizacích pomocí např. opět Metropolisova
algoritmu. Tím je zajištěna přítomnost všech teplot žíhání
v každém okamžiku optimalizace a parametry algoritmu jsou tak nezávislé na
problému i datech. Velikost náhodných změn strukturního modelu je
větší pro vyšší teploty a může být automaticky kontrolována
např. předem daným pevným poměrem mezi přijatými “lepšími“
a “horšími“ modely v Metropolisově algoritmu (obr. 3).
Někdy se
v krystalografické literatuře setkáme s pojmem “Reverse Monte
Carlo“, který je užíván některými autory [43] pro aplikaci tohoto
optimalizačního algoritmu na neuspřádané struktury a kapaliny
(modelování Pair Distribution Function), ale i na krystalické látky [40].
3. Vývojové teorie - genetický
algoritmus
Genetický algoritmus je založen
na Darwinově evoluční teorii. Pro historii jeho využití
v globální optimalizaci odkazujeme čtenáře na [44], jeho schema
je znázorněno na obr. 4.
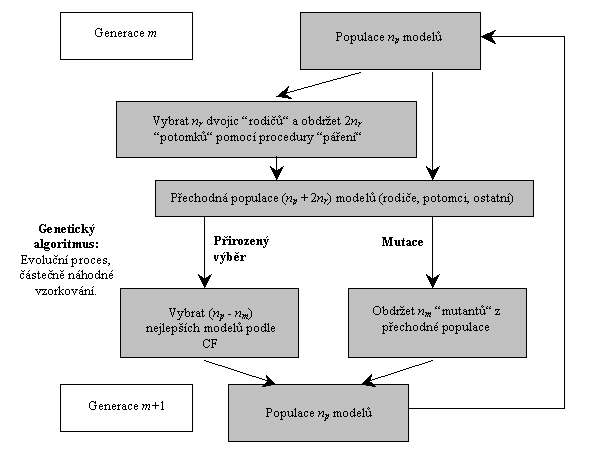
Obr. 4: Schema genetického algoritmu .
Každý strukturní model
z aktuální populace np modelů je popsán svým
“chromozomem“, který je souborem “genů“ – strukturních parametrů,
které určujeme. Charakteristickou každého modelu – jedince je jeho vhodnost
(fitness), popsána opět jednou CF nebo kombinací více CF. Aplikováním
genetických operátorů, “páření“ (kombinace genů obou
“rodičů“) a náhodné “mutace“, jsou vytvářeni noví jedinci a
jejich vhodnost je testována. Dle Darwinova principu o přežití
“nejlepších“ jedinců jsou formovány nové a nové generace; populace se
vyvíjí směrem ke globálnímu minimu hyperplochy CF. Tak jako
v simulovaném žíhání jsou kontrolnímí parametry algoritmu žíhací teplota a
velikost náhodných změn modelu, jsou v genetickém algoritmu
těmito parametry podíl a typ mutace na změně jedné generace,
způsob výběru “rodičů“ a typ jejich “páření“ a
velikost populace. Podíl mutace musí být dost výrazný, aby se zamezilo
“degeneraci“ genetického fondu (uvěznění v lokálním mimimu),
způsob výběru “rodičů“ může být např. “elitistický“.
Genetický algoritmus jako metodu globální optimalizace aplikovanou na
řešení struktur z práškových dat lze najít např. v [45, 46].
Jeho verze dle Lamarkianovy evoluční teorie [47] aplikuje také “vývoj“
jedince během jeho “života“ pomocí lokální minimalizace (posun jedince
k nejbližšímu lokálnímu minimu hyperplochy CF).
4. Další algoritmy
Řada dalších algoritmů
byla testována v globální optimalizaci aplikované na řešení struktur
z práškových dat. Jmenujme zde pouze algoritmus založený na sociálním
chování hejna jedinců, “swarm“, aplikovaný v [48] a velmi slibný hybridní
Monte Carlo algoritmus, aplikovaný v [49], který kombinuje výhody Monte Carlo
vzorkování hyperplochy CF s efektivností lokalizace jejích minim pomocí
formalismu molekulární dynamiky.
Který algoritmus je nejlepší?
Přímá odpověď neexistuje neboť srovnání není jednoduché a
závisí ve velké míře na problému, na který globální optimalizaci
aplikujeme. Simulované žíhání má však velkou výhodu oproti genetickému
algoritmu v menším počtu kontrolních parametrů algoritmu a
může být proto snadněji naprogramován (zvlástě ve své
podobě paralelního žíhání) jako “black-box“ algoritmus. Řada
pokusů byla také učiněna o kombinaci výhod obou algoritmů
v jeden hybridní [50].
Jak pomoci globální
optimalizaci?
Pravděpodobnost nalezení
globálního minima lze zvýšit omezením objemu elementární buňky, kterou
musí algoritmus vzorkovat (to odpovídá zjednodušení tvaru hyperplochy CF). Lze
použít např. informaci o molekulárních obálkách, metodu známou z krystalografie
proteinů (kde prášková difrakce našla řadu inspirací, protože se
potýká se stejným problémem, nízký poměr počtu dat k volným parametrům).
Molekulární obálky jsou vlastně Fourierovy mapy s velmi nízkým
rozlišením (periodické nodální plochy), zpočtené z několika nízkoúhlových
dobře rozlišených reflexí. Iso-plochy v takovéto mapě od sebe
oddělují oblasti s vyšší (polohy atomů) a nízkou (prázdné
oblasti) elektronovou hustotou. Algoritmus globální optimalizace pak prohledává
jen ty první. Aplikaci lze najít v [51].
Na začátku optimalizace je
strukturní model mnohem více vzdálen od pravdivého řešení než když se
algoritmus blíží globálnímu minimu. Tuto informaci lze do algoritmu vložit
pomoci pravděpodobnosti (likelihood), že takový model bude odpovídat pozorovaným
difrakčním (či jiným) datům. Hrubě řečeno
modifikujeme algoritmus tak, aby byl více “tolerantní“ ke špatným modelům
a nebral je příliž “vážně“. Myšlenku lze rozšířit tak, že
produkujeme úmyslně špatný model tím, že je jenom částečný
(vynecháme např. molekulu rozpouštědla). Takový model je pak menší
(méně volných parametrů) a hyperplocha CF jednodušší. Chybějící
část modelu (blur) a její příspěvek do difrakce však musí být
správně popsán, právě pomocí maximum likelihood formalismu. Aplikaci
lze najít v [52].
Poslední příspěvek,
výpočetní program FOCUS, vlastně nepatří do metod globální
optimalizace strukturního modelu, ale zařazujeme ho zde, protože využívá
také “inteligentní“ vzorkování přímého prostoru (elementární buňky).
Je to metoda, která pracuje s integrálními intensitami obdrženými
s práškového difrakčního záznamu a používá dodatečnou chemickou
informaci (znalost typických topologií, atomových koordinací, příbuzných
krystalových struktur) pro interpretaci a vzorkování Fourierových map
elektronových hustot. Při nalezení “slibného“ fragmentu v mapě
je celý cyklus opakován s fázemi reflexí obdrženými z tohoto
částečného strukturního modelu až do identifikace modelu schopného
již vypřesňování. Aplikaci na krystalografii zeolitů lze najít v
[53].
Závěr
Zde končí etapa řešení
strukury, na kterou navazuje vypřesnění struktury prováděné dnes
téměř výhradně pomocí Rietveldovy metody a analýzy Fourierových
map. Jak je vidět z obr. 1 neexistuje jediná “zlatá“ cesta
v bludišti řešení krystalových struktur pomocí práškové difrakce.
Výkonnost metod pracujících v reciprokém a v přímém prostoru je dnes
srovnatelná jak pro molekulární krystaly, tak i pro látky neobsahující
jasně definované molekuly (extended crystals, inorganické látky).
Větší úspěšnost jedné nebo druhé metody je silně podmíněna
daným problémem. Vývoj obou typů metod stále intenzivně
pokračuje, je zajímavé podotknout, že úspěch globální optimalizace
není dán jenom vzrůstem výpočetní výkonnosti, ale ve velké míře
i vývojem lepších algoritmů.
Ve spěchu aplikace dnešních
výkonných metod práškové difrakce je však nutno nezapomenout na první kroky
v bludišti obr. 1, vzorek a měření dat. Více času stráveného
na syntéze naší látky produkující vzorek bez příměsí a s užšími
difrakčními liniemi jakož i na optimalizaci difrakčního experimentu
se nám bohatě vrátí ve snazší analyze dat. Staré přísloví stále platí:
“práce kvapná, málo platná“.
Odkazy
[1] David W.I.F, Shankland
K., McCusker L.B. & Baerlocher Ch., Editors; Structure Determination
from Powder Diffraction Data, IUCr. Monographs on Crystallography 13,
Oxford University Press, 2002
[2] Visser, J.; J. Appl.
Crystallogr. 2 (1969) 89-95
[3] Boultif, A. & Loüer, D.;
J. Appl. Crystallogr. 24 (1991) 987-993
[4] Werner, P.-E., Eriksson, L. &
Westdahl, M. ; J. Appl. Crystallogr. 18 (1985) 108-113
[5] Rietveld, H.M.; J. Appl.
Crystallogr. 2 (1969) 65-71
[6] Le Bail, A.; Accuracy in
Powder Diffraction II, Proceedings of the International Conference,
May 26-29, 1992 NIST Special Publication 846, E. Prince and J.K. Stalick, Editors
page 213.
Také: http://www.cristal.org/iniref/lbmeth.html
[7] Pawley, G.S.; J.
Appl. Crystallogr. 14 (1981)
357-361
[8] Markvardsen, A., David,
W.I.F., Johnson, J.C. & Shankland, K.; Acta Crystallogr. A57
(2001) 47-54
[9] Altomare, A., Foadi, J., Giacovazzo,
C., Guagliardi, A. & Moliterni, A.G.G.; J. Appl. Crystallogr. 29
(1996) 674-681
Také: http://www.irmec.ba.cnr.it/
[10] Jansen, J., Peschar, R. &
Schenk, H. ; J. Appl. Crystallogr. 25 (1992) 237-243
[11] David, W.I.F.; J. Appl.
Crystallogr. 20 (1987) 316-319
[12] Estermann, M.A. &
Gramlich, V.; J. Appl. Crystallogr. 26 (1993) 396-404
[13] Zachariasen, W.H. &
Ellinger, F.H.; Acta Crystallogr. 16 (1963) 369-375
[14] Shankland, K., David, W.I.F.
& Sivia, D.S.; J. Mater. Chem. 7(3) (1997) 569-572
Také: Brunelli, M.,
Wright, J.P., Vaughan, G.B.M., Mora, A.J. & Fitch, A.N.; Angew. Chem.
Int. Ed. 42 (2003) 2029-2032
[15] Dahms, M. & Bunge,
H.J.; Textures and Microstructures 6
(1986) 167-179
Také: Bunge, H.J., Dahms,
M. & Brokmeier, H.G.; Cryst. Rev.
2 (1989) 67-88
Také: Hedel, R., Bunge,
H.J. & Reck, G.; Textures and
Microstructures 29 (1997)
103-126
[16] Wessels, T., Baerlocher, Ch.,
McCusker, L.B. & Creyghton, E.J.; J. Am. Chem. Soc. 121
(1999) 6242-6247
Také: Wessels, T.,
Baerlocher, Ch. & McCusker, L.B.; Science 284 (1999) 477-479
Také: http://www.kristall.ethz.ch/LFK/research/zeolite_group/Texture/Texture.html
[17] Černý, R.; Advances in X-ray Analysis 40 (1998) CD-ROM
Také: Černý, R. Mat.
Sci. Forum 321-324 (1999) 22-28
Také: Černý, R. Mat.
Sci. Forum 378-381 (2001) 24-29
Také: http://crystsun1.unige.ch/cerny/rcerny.htm
[18] Rius, J.; Acta Crystallogr.
A49 (1993) 406-409
[19] Bricogne, G.; Acta
Crystallogr. A47 (1991) 803-829
[20] Gilmore, C.J. &
Nicholson, W. ; Trans. Amer. Cryst. Association. 30 (1994)
15-27 Také: http://www.chem.gla.ac.uk/staff/chris/powder.htm
[21] Shankland, K., David, W.I.F.
& Csoka, T.; Z. Kristallogr. 212 (1997) 550-552
[22] GULP(General
Utility Lattice Program)
Také: http://www.ch.ic.ac.uk/gale/Research/gulp.html
[23] Engel G.E., Wilke S., Knig
O., Harris K.D.M. & Leusen F.J.J.; J. Appl. Cryst. 32 (1999)
1169-1179
Také: http://www.accelrys.com/cerius2/powder.html
[24] TOPAS User’s Manual, Bruker
AXS GmbH, 2000
Také: Coelho, A.A.; J.
Appl. Cryst. 33 (2000) 899-908
[25] Putz H., Schön J.C. &
Jansen M.; J. Appl. Cryst. 32 (1999) 864-870
Také: http://www.crystalimpact.com/endeavour/
[26] Kariuki B.M.,
Serrano-Gonzáles H., Johnston R.L. & Harris K.D.M.; Chem. Phys. Lett.
280 (1997) 189-195
[27] Andreev Y.G., Lighfoot, P.
& Bruce P.G.; J. Appl. Cryst. 30 (1997) 294-305
[28] Floudas, C.A.; Deterministic
Global Optimization: Theory, Algorithms and Applications, Kluwer, Dordrecht
1999.
Také: http://www.princeton.edu/~chemical/faculty/floudas/floudas.html
[29] Catlow, C.R.A., Cormack, A.N.
& Theobald, F. ; Acta Crystallogr. B40 (1984) 195-200
[30] Deem, M.W. & Newsam,
J.M.; Nature 342 (1989) 260-262
[31] Taylor, J.C. & Wilson,
P.W.; Acta Crystallogr. B30 (1974) 2664-2667
[32] Chernyshev,
V.V. & Schenk, H.; Z. Kristallogr. 213 (1998) L1-L3
[33] Masciocchi, N., Bianchi, R.,
Cairati, P., Mezza, G., Pulati, T. & Sironi, A.; J. Appl. Cryst. 27
(1994) 426-429
[34] N. Metropolis, A.W. Rosenbluth, M.N. Rosenbluth, A.H. Teller and
E. Teller: J.Chem. Phys. 21
(1953) 1087-1092
[35] Harris, K.D.M., Tremayne, M., Lightfoot, P. & Bruce, P.G.; J.
Am. Chem. Soc. 116 (1994) 3543-3547
[36] Andreev Y.G., MacGlashan G.S. & Bruce P.G.; Phys. Rev B,
Cond. Matter 55 (1997) 12011-12017
[37] David W.I.F.,
Shankland K. & Shankland N.; J. Chem Soc. Chem. Commun. 8
(1998) 931-932
Také: http://www.ccdc.cam.ac.uk/prods/dash/#ref1
[38] Pagola, S., Stephens, P.W.,
Bohle, D.S., Kosar, A.D. & Madsen, S.K.; Nature 404 (2000)
307-310
Také: http://powder.physics.sunysb.edu/programPSSP/pssp.html
[39] Favre-Nicolin, V. & Černý, R.; J.
Appl. Cryst. 35 (2002) 734-743
Také:
http://objcryst.sourceforge.net/
[40] Le
Bail, A; Materials Science Forum 378-381(2001) 65-70
Také: http://www.cristal.org/sdpd/espoir/index.html
[41] Toraya, H. & Yamazaki, S.; Acta Crystallogr. B58 (2002) 613-621
[42] Falcioni M. & Deem. M.W.; J. Chem.
Phys. 110 (1999) 1754-1766
Také: http://ccp14.semo.edu/ccp/web-mirrors/zefsa/zefsaII/
[43] Tucker,
M.G., Dove, M.T. & Keen, D.A.; J. Appl. Cryst. 34 (2001)
630-638
Také:
http://www.studsvik.uu.se/Software/rmc/rmcpow.htm
[44] Michalewicz,
Z.; Genetic Algorithms + Data Structures = Evolution Programs,
3rd edn., Springer-Verlag, Berlin 1996
[45] Shankland K., David W.I.F.
& Csoka T.; Z. Kristallogr. 212
(1997) 550-552
[46] Kariuki B.M.,
Serrano-Gonzáles H., Johnston R.L. & Harris K.D.M.; Chem. Phys. Lett.
280 (1997) 189-195
Také: http://www.dur.ac.uk/j.m.hutson/ccp6-98/node36.html
[47] Turner, G.W., Tedesco, E.,
Harris, K.D.M., Johnston, R.L. & Kariuki, B.M.;
Chem.
Phys. Letters 321 (2000) 183-190
[48] Csoka,
T. & David, W.I.F.; Acta Crystallogr. A55 (1999) Supplement
[49] Johnston
J. C., David W. I. F., Markvardsen A. J. and Shankland K.; Acta Cryst. A
58 (2002) 441-447
[50] http://www.densis.fee.unicamp.br/~moscato/memetic_home.html
Také: http://bruce.edmonds.name/aigp/
[51] Brenner
S., McCusker L. B. & Baerlocher Ch.; J. Appl. Cryst. 35
(2002) 243-252
Také:http://www.kristall.ethz.ch/LFK/research/zeolite_group/PNS/Structure_Envelopes.html
[52] Markvardsen
A.J, David W.I.F. & Shankland K.; Acta
Cryst. A58 (2002) 316 – 326
[53] Grosse-Kunstleve R.W., McCusker L.B. &
Baerlocher Ch.; J. Appl. Cryst. 30 (1997) 985-995
Také: http://www.kristall.ethz.ch/LFK/research/zeolite_group/FOCUS/FOCUS.html