COMBINATION OF MODELING AND EXPERIMENT IN ANALYSIS OF
PARTIALLY DISORDERED STRUCTURES.
Pavla Čapková1,
Jaroslava Repáková1, Bohdan Koudelka1, Miroslav Pospíšil1, Miroslava Trchová2, Zdeněk Weiss3, Vítězslav Zima2
and Michal Ilavský2
1Faculty of
Mathematics and Physics Charles University Prague, Ke Karlovu 3, CZ-12116 Prague, Czech Republic; E-mail:
capkova@karlov.mff.cuni.cz ; 2Institute of Macromolecular Chemistry,
Academy of Sciences of the Czech Republic, 16206 Prague 6, Czech republic; 3Technical
University Ostrava, 70833 Ostrava, Czech Republic;
Abstract:
Strategy of structure analysis for partially
disordered crystal structures has been worked out, based on combination of
modeling (i.e. force field calculations) and experiment (diffraction methods
and vibration spectroscopy). Modeling in conjunction with experiment enables us
to analyze the disordered structures, where the conventional diffraction
analysis fails. Experiment plays a key role in modeling strategy and in
corroboration of modeling results. X-ray powder diffraction and IR spectroscopy
were found as very useful complementary experiments to molecular modeling.
Molecular mechanics and molecular dynamics simulations were carried out in Cerius2/Materials Studio
modeling environment. An overview of structures solved by this method will be
presented, especially intercalates, liquid crystals and liquid crystalline
polymers.
Introduction
Intercalates, liquid crystals and liquid
crystalline polymers belong to partially disordered structures, where the
disorder obstructs the conventional structure analysis based on diffraction
method only. Due to the disorder it is also impossible to prepare the single
crystals of reasonable size for the diffraction measurements and powder
diffraction pattern is in addition to the disorder affected by the preferred
orientation of crystallites. In such a case molecular modeling i.e. molecular
mechanics and molecular dynamics represent very powerful tool in structure
analysis of these systems. Molecular mechanics is a method of optimization of
the structure and bonding geometry by energy minimization, where the energy is
described by an empirical force field [1]. These force field calculations
enable us to determine the structure of supramolecular systems, which are too
large for ab-initio quantum chemical
calculations and which exhibit certain degree of disorder and consequently the
diffraction analysis fails in structure determination. Molecular dynamics
simulations introduce the control of temperature and pressure to the system and
enable us to study the dynamic processes like diffusion and phase transitions.
Classical molecular dynamics involves the calculation of the time dependent
movement of each atom in a molecule [1,2]. This is achieved by solving Newton's
equations of motion. The temperature and the distribution of atomic velocities
in a system are related through the Maxwell-Boltzmann equation [1,2].
Strategy of modeling
The strategy of modeling (i.e. the building of
the initial models, the set up of energy expression, the choice and test of the
force field, the definition of rigid fragments, the set up of fixed and
variable structure parameters etc.) should be based on the available
experimental data. X-ray powder diffraction a vibration spectroscopy has been
found as very convenient complementary experiments to the molecular modeling.
Comparison of x-ray powder diffraction patterns and IR/Raman spectra for the
host structure, guest compound and intercalate reveals the changes of the host
structure and guest species during intercalation and help to create the
modeling strategy [3].
All the computational methods searching for the
global energy minimum have to generate a large number of initial models using
three different ways [4]: (1) Deterministic method for generation of starting
models, performing the systematic grid search that covers all areas of the
potential energy surface; (2) Molecular dynamics generating the starting
geometry; (3) Stochastic methods /Monte Carlo, Genetic Algorithm/. In modeling
of intercalated structures we used first two methods: the grid search using
program SUPRAMOL [4] with the final
energy minimization in Cerius2
and molecular dynamics in Cerius2
[5]. The first method, performing the systematic grid search can be used easily
to generate starting models in case of small organic guest molecules or almost
rigid large guests and rigid host layers. On the other hand molecular dynamics
is very convenient method for generation of starting models in case of large
flexible organic guest molecules.
As a result of molecular modeling we can get
the structure model, the energy characteristics (energy of intermolecular
interactions and charge distribution) and disorder characteristics (the type
and degree of the disorder). Molecular simulations enable the prediction of
structure and properties and thus represent big challenge for the design of new
materials with desirable properties. All results presented in this paper were
obtained using Cerius2/Materials Studio modeling environment from
(MSI/Accelrys) [5].
Intercalated
layered structures
Intercalation means an insertion of a guest molecule or ion into a
suitable crystal structure without major rearrangement of the solid host
structure [6,7]. Intercalation requires that the host structure has a strong
covalent network of atoms, which remains unchanged on the intercalation
reaction and that there are vacant sites in the structure. These vacant sites
should be interconnected and of suitable sites to permit the diffusion of the
guest species into the host structure. Layered crystal structures satisfy these
requirements very well being able to accommodate very large guest molecules in
the interlayer space by the free adjustment of the interlayer separations. A
strong intralayer and weak interlayer bonding characterizes layered structures.
A survey of different types of layered host structures and discussion of
intercalation reactions for neutral and charged layers is given in [6,7].
Intercalation provides new routes for the synthesis of materials with
controlled changes in the chemical and physical properties.
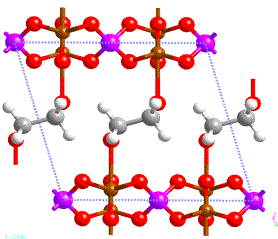
Figure 1:
High temperature phase of vanadyl
phosphate intercalated with dioxane
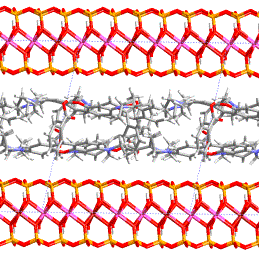
Figure 2: One example of intercalated layered silicates; [8] Montmorillonite intercalated with rhodamine B. Anchoring of dye
molecules on silicates layers changes their optical activity, (absorption
spectra, photoluminiscence etc. ). The way of guest anchoring to the host
layer, the conformation changes, the arrangement of guest molecules in the
interlayer space (monomers, dimers and higher aggregates) represent crucial
parameters for optical properties of the intercalate.
These properties can be tuned by the proper choice of the host-guest
combination, by the guest concentration and by co-intercalation of further
guest species. This allows creating a large variety of structures for a wide
scale of practical use. Intercalates can be used as adsorbents, catalysts,
pharmaceutical products, chemical sensors, ionic conductors and various kind of
electrochemical and opto-electronical devices [7].
Intercalation is in fact an
insertion of a known molecule into a known crystal structure, consequently the
structure analysis of intercalates has to solve specific problems. That
means: to find the positions,
orientation and ordering of the guest molecules in the host structure, to find
the way of layer stacking, conformational changes of guest molecules in the
interlayer space of the host structure and to characterize a possible disorder
in guest arrangement and consequently in the layer stacking.
Combination of modeling with X-ray and
synchrotron powder diffraction and with IR/Raman spectroscopy has been used to
solve the structure of following series of intercalates: (1) layer silicates
intercalated with organic and inorganic species (sorbents, catalysts,
photo-function units), (2) graphite intercalated with transition metal
chlorides (two-dimensional conductors), (3) tantalum sulfide intercalated with
dye molecules (superconductors), and
(4) vanadyl and zirconium phosphate intercalated with small organic molecules
(catalysts, chemical sensors, proton conductors).
Structural
phase transitions in liquid crystals and liquid crystalline polymers:
Liquid crystalline polymers can be obtained from specific monomers
containing a mesogenic rigid segment. However the main trouble of processing
such polymers is the high melting point, sometimes exceeding the decomposition
temperature. To decrease the phase transition temperature, the flexible chain
spacers between rigid mesogenic segments are inserted [9]. Each modification of
monomers leads to the changes of thermal and mechanical properties of polymer
systems and therefore the investigation of the structure and properties of monomers
is important for understanding the of thermal and mechanical behaviour of
derived polymer systems.
Phase
transitions in molecular crystal of 4,4’- bis(6-hydroxy-1-hexyloxy)biphenyl
have been studied using molecular dynamics simulations to reveal the mechanism
of the phase transition into liquid crystalline state and to explain similar
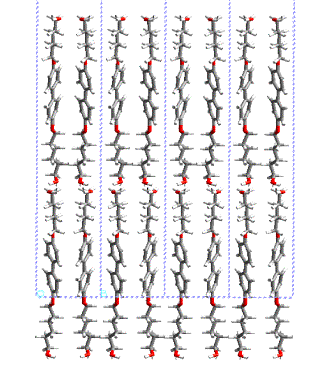
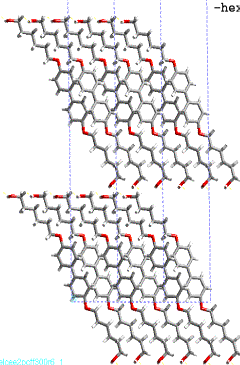
phase transition in polyuretane derived from this monomer. Figure 2a,b shows
the layered crystal structure of 4,4’- bis(6-hydroxy-1-hexyloxy)biphenyl. In
the figure 3 one can see the conformational changes of molecules as a result of molecular dynamic
simulations carried out for room temperature, for 400 K in liquid crystalline
state and for 500K above the melting point [11].
![]()
The
design of new polymers with desirable thermal behavior requires the search for
monomers with suitable combination of rigid mesogenic group and flexible
chains. One way to this goal is the use of suitable side chains in flexible
part of molecule. One example of this investigation is monomeric unit presented
in the figure 5, which has been used with diisocyanate as a copolymer to design
polyuretane with liquid crystalline behavior. Figure 6 shows the snapshots
extracted from dynamic trajectory file at 300K at various time steps in dynamic
trajectory, illustrating the rigidity of the polymeric chain in case of
polyuretane.
![]()
Figure
3: Layered crystal structure
of 4,4’- bis(6-hydroxy-1-hexyloxy)biphenyl; view along a and b axis.
The stiffness and rigidity of single polymer chain
was described using two parameters, end to end distance and radius of gyration.
The time dependence of these characteristics during dynamic simulations allows
us to characterize the thermal and mechanical behavior of polyuretane. Characteristics
obtained using dynamic simulation were in good agreement with results of
experiment.
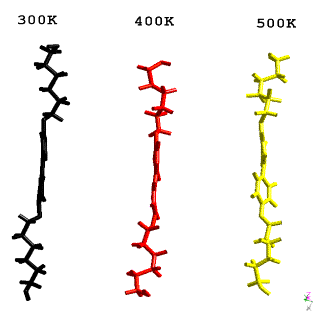
Figure 4:
Conformation of 4,4’-
bis(6-hydroxy-1-hexyloxy)biphenyl molecule:
at 300K, after the phase transition into liquid crystalline state at 400K,
when the distortions of flexible chains result in disordered layer stacking in
crystal structure in figure 2 and at 500K after the melting point, when the
distortion of the rigid part of molecule leads to the melting of crystal
structure.
![]()
Figure
5: The monomeric unit used
together with diisocyanate for the design of polyuretan. (n = 6, X = Phenyl).
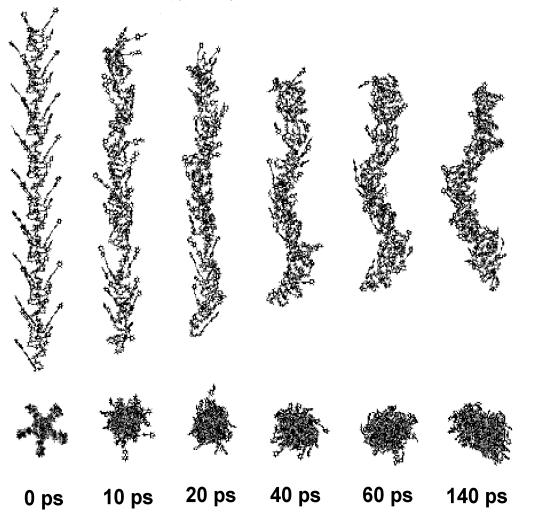
Figure 6: Snapshots extracted from dynamic trajectory at 300K, illustrating
the degree of deformation and rigidity of the single polyuretane chain composed
from diisocyanate and diol with side chain from the figure 5.
References:
[1] P. Comba, & T.W. Hambley
(Ed.): Molecular Modeling. Weinheim, New York,
1995 VCH Verlagsgesellschaft mbH.
[2] D. Frenkel & B.
Smit: Understanding Molecular Simulation. San Diego, New York, London, 1996,
Academic Press.
[3] P.
Čapková, M.Trchová, P. Hlídek, H. Schenk & M.Ilavský:, J. Molecular Structure, 559 (2001) 209-217.
[4] B. Koudelka & P. Čapková, J. Mol. Model, 8 (2002), 184-190.
[5] Cerius2 documentation, June 2000, Molecular simulation/Accelrys, Inc. San Diego,
USA
[6] A.J.
Jacobson: Intercalation reaction of layered compounds. In: Solid state
chemistry compounds, eds.: Cheetham A.K., Day P., Oxford (1992), Clarendon
Press.
[7] A.
Lerf: Intercalation Compounds in Layered Host Lattices: Supramolecular
Chemistry in Nanodimensions. In: Handbook of
Nanostructured Materials and Nanotechnology, vol. 5, eds.: Nalwa H.S,
Academic Press, New York, (2000).
[8] M.
Pospíšil, P. Čapková, H. Weissmannová, Z. Klika, M. Trchová, M. Chmielová &
Z. Weiss, J. Mol. Model, 9
(2003) 39-46.
[9]
M. Ilavský, K. Bouchal, H. Valentová, F. Lednický, A. Sikora & J. Baldrian
J.: Macromol Sci. Phys. B37 (1998) 645-666.
[10] K. Goubitz, P. Čapková, K. Melánová, W. Molleman H. Schenk, Acta Cryst., B57 (2001)
178-183.
[11] P.
Čapková, M. Trchová, P. Hlídek, H. Schenk, M. Ilavský: J. Mol. Str. 559 (2001)
209-217.