Binding of Peptide Deformylase to the Ribosome Surface Modulates Exit Tunnel Interior
Hugo McGrath, Michaela Černeková, and Michal H. Kolář
Department of Physical Chemistry, University of Chemistry and Technology, Technická 5, 16628 Prague, Czech Republic
Ribosomes are the biomolecular factories in charge of protein synthesis and are thus essential for life as we know it. In order to better understand proteosynthesis it is important to characterize the ways in which it is regulated. The regulatory mechanisms may involve conformational changes of the ribosome induced by external factors possibly transferred over large distances. The principles of this allosteric communication between distant ribosome parts are not fully understood yet. Computer simulations represent a convenient tool to study dynamic complex systems including the ribosome [1]. Here we investigate peptide deformylase, an enzyme that binds to the ribosome surface near the ribosomal protein uL22 during translation and modifies the emerging nascent chain, to understand how conformational motion of the ribosome is affected by external factors [2] (preprint: doi.org/10.1101/2022.04.20.488877).
We have performed all-atom molecular dynamics simulations of the entire ribosome. The simulated system consists of about 2 million atoms so for the microsecond-scale simulations of ours a supercomputing facility involving thousands of CPU cores had to be employed.
We analyzed these simulations using principal component regression, a form of supervised machine learning. The results indicate conformational changes of the ribosomal protein uL22 inside the ribosomal exit tunnel that are correlated with deformylase binding. This suggests a possible effect of the deformylase on the nascent peptide transport through the tunnel.
The research was supported by the Czech Science Foundation (projects 19-06479Y and 23-05557S). MČ acknowledges the support of the Internal Grant Agency of UCT Prague (project 403-88-2091). We also thank the Max Planck Computing and Data Facility.
1. Lars V Bock, Sara Gabrielli, Michal H Kolář, and Helmut Grubmüller. Simulation of complex biomolecular systems: The ribosome challenge. Annual Review of Biophysics, 52, 2023.
2. Hugo McGrath, Michaela Černeková, and Michal H Kolář. Binding of the peptide deformylase on the ribosome surface modulates the exit tunnel interior. Biophysical Journal, 121(23):4443–4451, December 2022.
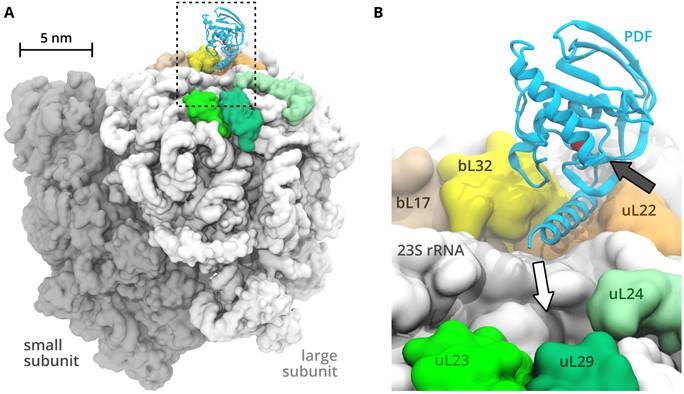
Figure 1: A: The ribosome with peptide deformylase bound to the surface. B: Detail of bound peptide deformylase.