On the Importance of Physically Correct Models for Protein-Ligand Binding
M. Lepšík1, A. Imberty2, P. Hobza1, J. Řezáč1
1 Institute of Organic Chemistry and Biochemistry (IOCB), Czech Academy of Sciences, Prague, Czech Republic 2 Centre de recherches sur les macromolécules végétales (CERMAV), CNRS, Grenoble, France
martin.lepsik@uochb.cas.cz
Understanding protein-ligand binding in atomistic details is key to success in structure-based drug design. I will discuss two recent approaches: i) effective electronic polarization scheme for classical molecular dynamics (MD)[1] and ii) parametrized semiempirical quantum mechanics (SQM)-based scoring function [2, 3]. The former method includes polarisation in classical additive force fields via charge scaling. We have shown its power in correct structural description of the bridging Ca2+ ions in lectin/carbohydrate complex (Fig. 1A, B) [1].
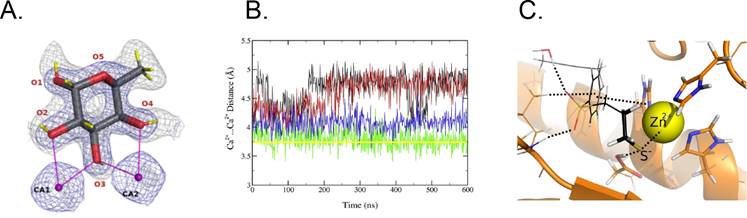
Figure 1. A. L-Fucose binding to two calcium ions in LecB lectin (PDB: 7PRG). B. Ca2+···Ca2+ distances from MD with standard Ca2+ parameters (black, red) and scaled-charge parameters (blue, green) compared with the crystal value (yellow). C. The active site of metalloprotein (brown ribbon) with Zn2+ (yellow) and thiolate (S-) group of ligand. Noncovalent interactions are shown as dotted lines.
A universal method, which includes also other quantum effects, such as charge transfer or σ-hole bonding, is quantum mechanics. We have developed an SQM-based scoring function which, due to its chemical generality, outperforms standard academic and commercial packages in sampling, ranking and virtual screening. In summary, developing and applying physically correct models of protein-ligand binding heads toward an unrivalled qualitative and quantitative enhancement of the predictive power of computer-aided drug design.
1. M. Lepsik, et al. Induction of rare conformation of oligosaccharide by binding to calcium-dependent bacterial lectin: X-ray crystallography and modelling study. Eur J Med Chem., 177, (2019), 212-220.
2. M. Lepšík, et al. The Semiempirical Quantum Mechanical Scoring Function for In Silico Drug Design. ChemPlusChem, 78, (2013), 921 – 931.
3. A. Pecina, et al., SQM/COSMO Scoring Function: Reliable Quantum-Mechanical Tool for Sampling and Ranking in Structure-Based Drug Design. ChemPlusChem, 85, (2020), 2362 –2371.
ML, PH and JŘ are supported by ChemBioDrug CZ.02.1.01/0.0/0.0/16_019/0000729 project from the European Regional Development Fund (OP RDE). ML acknowledges funding from the Horizon 2020 research programme under the Marie Sklodowska-Curie grant agreement No. 795605, E.U.