The benchmark of 31P NMR parameters in phosphate: a case study on structurally constrained and flexible phosphate
Jiří Fukal,ab Ondřej Páv,a Miloš
Buděšínský,a Jakub Šeberaa and Vladimír Sychrovskýac
aInstitute of Organic
Chemistry and Biochemistry, Academy of Sciences of the Czech Republic, v.v.i.,
Flemingovo náměstí 2, 166 10, Praha 6, Czech Republic. Email:
jiri.fukal@uochb.cas.cz
bCharles University, Faculty of Mathematics and Physics, Ke Karlovu
3, 121 16, Praha 2, Czech Republic
cDepartment of Electrotechnology, Electrical Engineering, Czech
Technical University, Technická 2, 166 27, Praha 6, Czech Republic
A benchmark for structural interpretation
of the 31P NMR shift and the 2JP,C NMR
spin–spin coupling in the phosphate group was obtained by means of theoretical
calculations and NMR measurements in diethylphosphate (DEP) and
5,5-dimethyl-2-hydroxy-1,3,2-dioxaphosphinane 2-oxide (cDEP). The NMR
parameters were calculated employing the B3LYP, BP86, BPW91, M06-2X, PBE0, KT2,
KT3, MP2, and HF methods, and the 6-31+G(d), Iglo-n (n = II, III), cc-pVnZ( n =
D, T, Q, 5), aug-cc-pVnZ( n = D, T and Q), and pcS-n and pcJ-n (n = 1, 2, 3, 4)
bases, including the solvent effects described with explicit water molecules
and/or the implicit Polarizable Continuum Model (PCM). The effect of molecular
dynamics (MD) on NMR parameters was MD-calculated using the GAFF force field
inclusive of explicit hydration with TIP3P water molecules. Both the optimal
geometries and the dynamic behaviors of the DEP and cDEP phosphates differed
notably, which allowed a reliable theoretical benchmark of the 31P
NMR parameters for highly flexible and structurally constrained phosphate in a
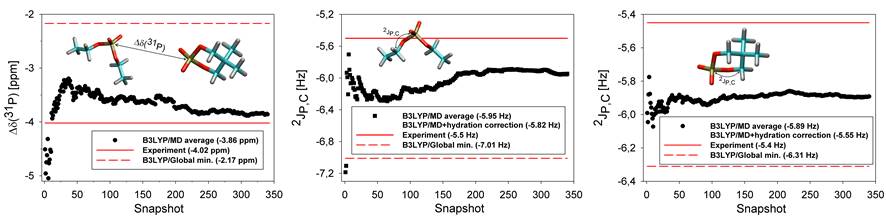
one-to-one relationship with the corresponding experiment. The calculated 31P
NMR shifts were referenced employing three different NMR reference schemes to
highlight the effect of the 31P NMR reference on the accuracy of the
calculated 31P NMR shift. The relative Δδ(31P)
NMR shift calculated employing the MD/B3LYP/Iglo-III/PCM method differed from
the experiment by 0.16 ppm while the NMR shifts referenced to H3PO4
and/or PH3 deviated from the experiment notably more, which
illustrated the superior applicability of the relative NMR reference scheme.
The 2JP,C coupling in DEP and cDEP calculated employing
the MD/B3LYP/Iglo-III(DSO,PSO,SD)/cc-PV5Z(FC)/PCM method inclusive of
correction due to explicit hydration differed from the experiment by 0.32 Hz
and 0.15 Hz, respectively. The NMR calculations demonstrated that reliable
structural interpretation of the 31P NMR parameters in phosphate must involve
both the structural and the dynamical components.