Theoretical Description of Carbohydrate-Aromatic CH-π Interactions Additive Properties via DFT and Ab Initio Calculations
S. Kozmon1,2, R. Matuška1,2, V. Spiwok3,
J. Koča1,2
1CEITEC – Central-European Institute of Technology, Masaryk University, Kamenice 753/5, 625 00, Brno, Czech Republic
2National Centre for Biomolecular Research, Faculty of Science, Masaryk University, Kamenice 5, 625 00, Brno, Czech Republic
3Institute of Chemical Technology, Department of Biochemistry & Microbiology, Technická 5, 166 28, Praha, Czech Republic
There are several ways how saccharides may interact with their receptors (e.g. classical hydrogen bonds, through metal ions as Ca(II)). The CH-π interactions that occur between carbohydrates and aromatic amino-acids are also strongly involved in carbohydrate-recognition process. However, their influence to the recognition process has been underestimated for a long time. Despite the fact, that CH-π interactions were recently proved to have strength (which means contribution to the recognition process) comparable to classical hydrogen bonds.
In previous study [1], we have introduced systematic DFT and high-level ab initio study of CH-π interaction features between benzene as the simplest representative of aromatic moiety in proteins and three saccharides – namely β-D-glucopyranose, β-D-mannopyranose and α-L-fucopyranose. Nevertheless, also condensated aromatic moieties as Trp residues are responsible for the CH-π-mediated recognition of carbohydrate molecules. Lutteke et al. [2] have shown, that Trp is the most common residue found in direct contact with β-D-glucopyranose molecules.
Introduced computational study [3] aims to describe the degree of additivity of the CH-π interaction analyzing the interaction energy of carbohydrate-benzene complexes with monodentate (one CH-π contact) and bidentate (two CH-π contacts) carbohydrate-naphtalene complexes. For illustration of carbohydrate-naphtalene complexes structure, see Figure 1A. All model complexes were optimized at DFT-D BP/def2‑TZVPP level of theory, followed by refinement of interaction energies at highly-correlated and accurate CCSD(T)/CBS level. Also Boltzmann-weighted populations of naphtalene-carbohydrate complexes were calculated for each carbohydrate apolar face (see Figure 1B).
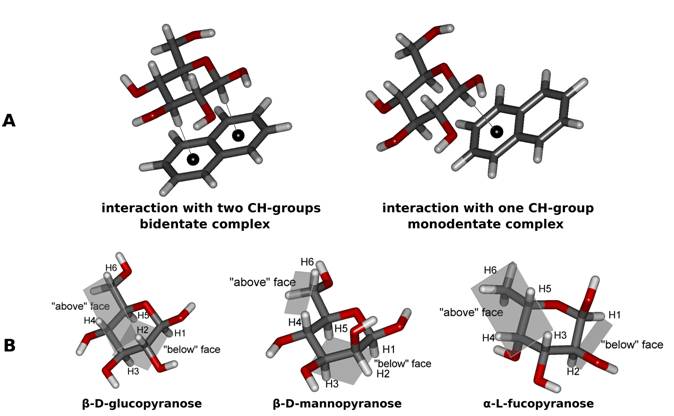
Figure
1: (A) Schematic
depiction of monodentate (one CH-π contact) and bidentate (two CH-π
contacts) carbohydrate-naphtalene complexes. (B) Structures and their apolar
faces involved in CH-π additivity study.
Bidentate carbohydrate-naphtalene complexes exhibit very high interaction energy values ranging from −7,15 kcal mol−1 to −10,79 kcal mol−1 for DFT-D level, and from −6,14 kcal mol−1 to −8,20 kcal mol−1 for CCSD(T)/CBS level. Values of interaction energy for bidentate carbohydrate-naphtalene complexes were compared with values for monodentate carbohydrate-naphtalene complexes and carbohydrate-benzene complexes.
The analysis unravels that the CH-π is not completely additive, because the interaction energy of bidentate complexes is higher (the interaction is weaker) than the sum of interaction energies of two corresponding monodentate complexes. However, deeper analysis discovers certain measurable degree of additivity. More precisely, the interaction energy of bidentate complex is 2/3 of the sum of interaction energies of appropriate monodentate complexes. Similarly, the interaction energy value for bidentante carbohydrate-naphtalene complexes is comparable to 4/5 of the sum of interaction energies of corresponding carbohydrate-benzene complexes.
Geometries of bidentate complexes are characteristic with shortening of H-π distance to the narrow range about 2,3 Å compared to monodentate complexes. The C-H bond line forms almost normal of the naphtalene plane with minimal deviations for all bidentate complexes. Taking into consideration calculated interaction energies, we may conclude that bidentate complexes of carbohydrates with condensed aromatic moiety (naphtalene) are very stable and rigid.
This study also serves as illustration that DFT-D methods describe CH-π interactions in qualitatively similar manner as more computationally demanding CCSD(T)/CBS method. Based on both performed studies, we may state that DFT-D approach may be utilized for computational treatment of larger complexes of biological interest, where CH-π dispersion interactions play non-negligible role.
This work was funded by the European
Community’s Seventh Framework Programme under
grant agreement no 205872, the Ministry of Education of the Czech
Republic (MSM0021622413, LC06030, MSM6046137305), and the Czech Science
Foundation (GD301/09/H004). The project is supported within the SoMoPro programme (project
No. 2SGA2747). The research leading to these results obtained financial
contribution from the European Union under the Seventh Framework Programme (FP/2007- 2013) by Grant Agreement No.
229603. The research is also co-funded by the South Moravian region. In
addition, this work was also supported within the project
‘‘CEITEC-Central European Institute of Technology’’
(CZ.1.05/1.1.00/02.0068) from European Regional Development Fund. The authors
would like to thank the Czech National Supercomputing Centre, MetaCentrum, for providing computational resources. Access
to the MetaCentrum computing facilities is provided
under the research intent MSM6383917201.
1. Kozmon S., Matuška R., Spiwok V., Koča
J., Chem. Eur. J., 17, (2011), 5680.
2. Lutteke T., Frank M.,
von der Lieth C. W., Nucleic Acids Res.,
33, (2005), D242.
3. Kozmon S., Matuška R., Spiwok V., Koča J., Phys. Chem. Chem. Phys., 13, (2011), 14215.