Intermolecular interactions displayed by X-ray diffraction
J. Hašek1, J. Dušková1, T. Skálová1, T. Koval2 and J. Dohnálek1,2
1Institute of Macromolecular Chemistry, Academy of Sciences of the Czech Republic, v.v.i., Heyrovského nám. 2, 162 06 Praha 6, Czech Republic
2Institute of Physics, Academy of Sciences of the Czech Republic, v.v.i., Na Slovance 2,182 21 Praha 6, Czech Republic,
hasek@imc.cas.cz
X-ray diffraction methods have been developed in a user-friendly and relatively quick method for determination of macromolecular structure, for analysis of molecular dynamics and for determination of reaction paths taking place in these systems. In principle, the method has not any limitations as far as the complexity of the bio-macromolecular adducts. The results are fully based on experiment (no a priory assumptions on structure neither molecular modeling are required) and thus they are very reliable in comparison with any other method. Accuracy of atom positions depends always on quality of the local electron density, but for typical data measured at 1.8 Å resolution the expected accuracy is about 0.1 Å.
Protein crystal can be defined as a regular three-dimensional scaffold of
protein molecules stabilized in solution by weak intermolecular forces. “The crystals”
are in hydrodynamic equilibrium with solvent and must be measured in solution.
Insensitive changes in solvent composition or solvent contents in the crystal quickly
destroy diffraction quality of the sample.
Protein crystals can exist in various concentrations (protein contents
from 15 % to 75 %). However, for each molecular stacking, there is only a
narrow range of solvent contents and the ingredient concentrations under which
the stacking preserves its periodicity.
X-ray structure analysis is a very reliable method and thus if one
observes adhesion between macromolecules, it is a definite prove that this
interaction exists under conditions present in the measured sample. However, because
of large surface of macromolecules, each protein has several different
interaction modes with preferences dependent on the solvent composition. Thus,
the diffraction methods can show as a rule several complexes between two
proteins with different affinity and stability. All the observed adhesion modes
are real under given circumstances. The decision which adhesion mode dominates
in the biological process in the cell may be simple or complex, but the
preparation of several crystal forms showing all adhesion modes is desirable to
get full view of the bio-processes in any case.
The real adhesion mode dominating in the bioprocess under investigation can
be different from the adhesion modes observed in solution because of many
competitive intermolecular interactions and spatial restrictions in the living cell.
Even more, the biologically relevant adhesion mode need not be the most stable
in solution, because the proteins are often oriented by anchoring in membrane
or are simply temporary complexed with other molecules forming the respective
organelle of the cell. Thus, many unknown factors can change the adhesion properties
of the inspected macromolecule in the bioprocess under investigation.
X-ray structure analysis is optimal tool for inspection of these
phenomena, because the crystalline samples studied have very similar
concentration of the protein material as it is in organelles of the living cell
(see Figure 1). The talk explains how “protein surface modifying agents PSMA” [1, 2] allow growing different
crystal forms showing different adhesion modes between proteins to get as far
as possible a complete review of all adhesion modes of the protein studied. It
also explains a role of PSMA in increasing the accuracy of the experimentally
determined atom positions under the desirable limit 0.1 Å (see Figure 2).
Applications of polymer materials as “protein surface modifying agents” and
behaviour of hydrophilic polymers in the bio-environment are summarized in [3].
The practical usage of these considerations in development of new
crystallization screens were published in [4]. It is expected, that the new
more complex usage of the X-ray diffraction methods as described here can
provide more realistic insight on the real behaviour of molecules in their
natural bio-environment.
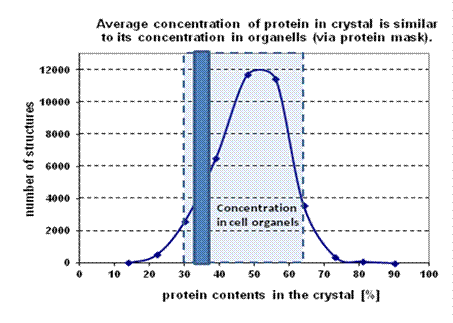
Figure 1. The graph shows a water contents in bio-macromolecular structures deposited in the PDB until
2012. It shows that a regular stacking of macromolecules into protein crystal
is not possible when protein concentration in “the solid phase” is lower than
30 % or higher than 70 %. To compare it with a standard biological system: the
dark rectangular area shows an average concentration of all biomolecules
in cells and the lightly coloured area shows their expected concentrations in
cell organelles.
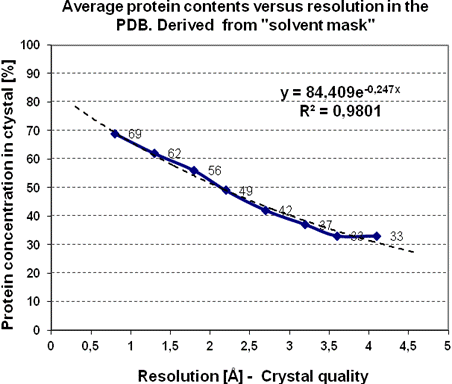
Figure 2. A higher content of water in the measured protein crystal leads on average to numerically higher “resolution” (to lower amount of diffraction data). The “protein surface modifying agents PSMA” can help to stabilize the macromolecular stacking in regular lattice and to get the resolution under the desirable limit 1.8 Å corresponding roughly to the accuracy of atom positions about 0.1 Å on average [3, 4].
The
work on this project was supported by the Grant Agency of the Czech Republic,
project no. 305/07/1073, P302/11/0855 and Grant Agency of the Czech Academy of
Sciences IAA500500701.
1. Hašek, J. Zeitschrift fur Kristallografie, 2006, 23, 613-618.
2. Hašek, J. Journal of Synchrotron Radiation, 2011, 18, 50-52.
3. Hašek, J. ,
Skálová, T., Dušková, J., Kolenko, P., Koval, T.,
Dohnálek, J. Crystallography Reviews,
2012, in press.
4. Skálová, T., Dušková, J.,
Hašek, J., Kolenko, P., Štěpánková, A, Dohnálek, J. J. Appl.Crystallogr., 2010, 43,
737-742.