A STRUCTURAL MODEL OF HUMAN MT2 MELATONIN RECEPTOR AND ITS MELATONIN RECOGNITION SITE
Ž. Sovová1, P. Mazna2,
J. Teisinger2, D.Štys1, T. Obšil2 and R.
Ettrich1
1Laboratory of
High Performance Computing, Institute of Physical Biology USB and Institute of
Landscape Ecology AS CR, University of South Bohemia, Zámek 136, CZ-373 33 Nové
Hrady, Czech Republic, email: ettrich@greentech.cz
2Institute of
Physiology, Czech Academy of Sciences, Vídeňská 1083, CZ-142 20 Prague
Keywords:
Molecular modeling, 3D-structure, membrane
proteins, melatonin receptor, melatonin
Abstract
Homology modeling of the hMT2 melatonin
receptor is reported. The deduced amino acid sequence shows high homology with
bovine rhodopsin, whose tertiary structure has been solved at 2.6 Å resolution
by X-ray crystallography. Docking of melatonin into the receptor site of the
protein structure was explored. The resulting structure contains seven putative
transmembrane domains connected by three extracellular and three intracellular
loops.
We have identified that for high-affinity
melatonin binding to hMT2 receptor are essential Val 204 and Leu 272 in
transmembrane domains five and six respectively as well as Tyr 298 in
transmembrane domain seven. We have also demonstrated the importance of Gly 271
for high-affinity melatonin binding to the hMT2 melatonin receptor
The pineal hormone
melatonin, is present in all vertebrate species including humans. Aside from
being an important regulator of seasonal reproduction and circadian rhythms
melatonin was reported to be potentially important immunomodulator, powerful
free radical scavenger and exerts oncostatic activity. Melatonin binding to
specific G protein-coupled receptors (GPCRs), designated as MT1, MT2 and Mel1c,
modulates wide range of intracellular messengers mediating hormone effects. MT1
and MT2 subtypes are expressed in mammals whereas Mel1c subtype has been cloned
from lower vertebrates (reviewed in [1] and [2]).
GPCRs contain seven
putative transmembrane domains connected by three extracellular and three
intracellular loops. It is widely accepted that TMs are involved in specific
interactions with ligand. Still, very little is known about actual arrangement
of TMs in majority of GPCRs, as except the light receptor rhodopsin [3]
structures of GPCRs at the atomic level are unknown.
Thus, the absence of
detailed structure of second mammalian melatonin receptor led us to
construction of three-dimensional model of the helical part of human MT2 (hMT2)
receptor generated by homology to the known crystal structure of the bovine
rhodopsin determined at a 2.6 Å resolution [3].
The choice of the
templates was restricted to the bovine rhodopsin, whose tertiary structure has
been solved at 2.6-Å resolution by X-ray crystallography and for which
the PDB coordinates were available [3]. The structure (1L9H) was extracted from
the Brookhaven Protein Data Bank (www.pdb.org) and loaded into SwissPdbViewer
[4], where we extracted a construct containing only one monomer. The primary
structures were aligned with by CLUSTALX [5].
The slow–accurate
mode with a gap opening penalty of 10 and a gap extensions penalty of 0.1 for
the local alignment was used as well as the Gonnet 250 protein weight matrix
and hydrophobic penalties for the amino acids GPSNDQEKR. The alignment used for
further modeling is shown in figure 1.
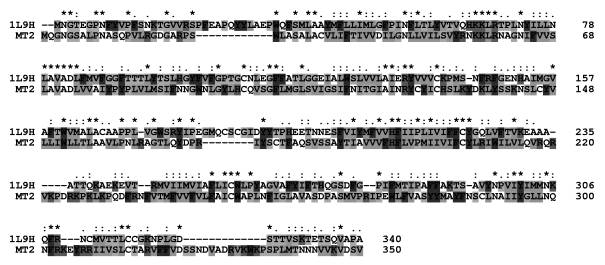
Fig. 1. Sequence alignment of
the MT2 melatonin receptor and bovine rhodopsin. Identical and similar amino
acids of the stronger groups are indicated with an asterisk and colon,
respectively. These amino acids should conserve the structure with a
probability of 95%. Dots indicate
similar amino acids of the lower groups that should conserve the structure with
a lower probability.
Three-dimensional
models comprising all non-hydrogen atoms were generated by the MODELLER6
package. [6] This is based on a distance restraint algorithm, satisfying
spatial constraints extracted from the alignment of the known protein, which is
the template structure, with the target sequence and from the CHARMM-22
force-field [7]. A bundle of five models from random generation of the starting
structure was calculated. The resulting models showed MODELLER target function
values of 2403, 2446, 2530, 2673, 2451, respectively. All models obtained were
subjected to a short simulated annealing refinement protocol available in
MODELLER. The tertiary structure models were checked with PROCHECK [8]. It
produces a Ramachandran diagram and allows examination of various structural
features such as bond lengths and angles, secondary structures and exposure of
residues to the solvent.
The structure of the
melatonin was built with the sketcher module of of InSight II, v2000.1,
(Accelrys Inc., San Diego, CA, USA) and geometry-optimized using the DISCOVER
force field cvff. To obtain the receptor-ligand complex the melatonin was
manually fitted into the binding site of the receptor. The starting point for
ligand docking was the orientation proposed by Grol and Jansen [15]. The ligand
was positioned by avoiding severe steric overlap with the receptor, trying to
keep the aromatic part of the melatonin close to the hydrophobic side chains.
The resulting complex was minimized in vacuo using SANDER with the ff99
forcefield included in AMBER 7.0 [9].
The quality of the
alignment can be seen as the most important step in homology modeling.
Therefore, the degree of similarity between the target sequence and the
template and the reliability of the alignment are the most critical problems.
These two problems are of course partially interconnected, since the degree of
similarity of two structures decreases with the degree of sequence identity
[10]. In our case the pair wise identity with bovine rhodopsin was quite low,
just about 21 %. However, the similarity between both sequences of about 48 %
was relatively high and makes homology modeling possible. Similarity in this
case included not only identical amino acids, but also indicated that amino
acids of the stronger groups were conserved. Stronger groups are: CSTA, NEQK,
NHQK, NDEQ, QHRK, MILF, HY, FYW. These amino acids should conserve the
structure and are marked in the alignment with two stars. For such a degree of
similarity, alignment errors were possible [11]. One basis of homology modeling
is the assumption that it is possible to define a unique optimal sequence-based
alignment that coincides with a structure-based alignment. This is not true in
general because every alignment program tries to maximize the number of
alignable residues, although these residues might not be spatially
superposable. This limitation and source of error is intrinsic and should
always be taken into account when estimating the degree of confidence of a
certain model.
The best Ramachandran
plot of the predicted structures calculated with PROCHECK, shown in Fig.2,
revealed a good quality stereochemistry, as indicated by the torsion angles F and Y.
The F, Y torsion angles of
85.1 % of the residues had values within the most favored areas and 10.8 % of
the residues had values within additionally allowed regions of the Ramachandran
plot. Six residues (1.9 %) were found in disallowed regions. This is acceptable
for a structure based on a template of 2.6 Å that has 3.1 % of its
residues either in generously allowed or even disallowed regions. The overall g
factor of the best structure obtained showed a value of –0.18. The g factor
should be above –0.5 and values below –1.0 may need investigation. To summarize
we can say that the final model gave the best results in all three categories.
It has the lowest MODELLER objective function, the highest percentage of
residues in the most favored regions and the highest g-factor. The g-factor of
our structure is only slightly lower than of the template structure (0.06)
which is an ideal result and shows that our MT2 structure fulfills all criteria
of a good quality model. It must be added that all five calculated models
differ only slightly in all done checks, which demonstrates the consistence of
the method that should ideally give identical results for every run. The model
structure placed into the membrane is shown in figure 2.
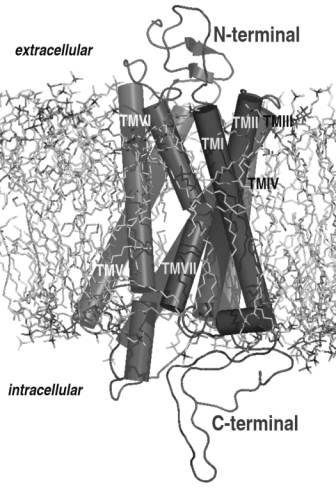
Fig. 2. The final model of the
MT2 melatonin receptor. The protein structure is placed into a lipid bilayer
system with 128 POPC lipids that was preequilibrated.
The model of the melatonin receptor consists
of seven membrane helices and 6 loops (Three intracellular and three extracellular).
Some of the helices are interrupted, which might point to regions of low
resolution. The interrupted helixes are: TM V (between Phe194 and Phe196), TM
VI (Arg235 to Thr239) and TM VII (Ala284 to Leu290).
Whereas the intracellular loops play a crucial
role in the receptor function, the role of the extracellular loops seems to be
marginal. Two loops seem to play the key role in the receptor function: the
small intracellular loop between helices 3 and 4 (Cys143 to Ser153) and the
longer loop between helices 5 and 6 (Leu226 to Pro240). Four helices form the
receptor site for melatonin, these are TM III (Tyr97 to Ile 129), TM V (Ser185
to Ile212), TM VI (Lys 228 to Ser263) and TM VII (Trp275 to Cys298).
The various
membrane-bound receptors the C-terminal plays an important role in regulation
the receptor function [12]. Also in our case the C-terminal (Asn301-Val350)
seems to have a rigid self supporting structure and therefore a functional
importance might be predicted.
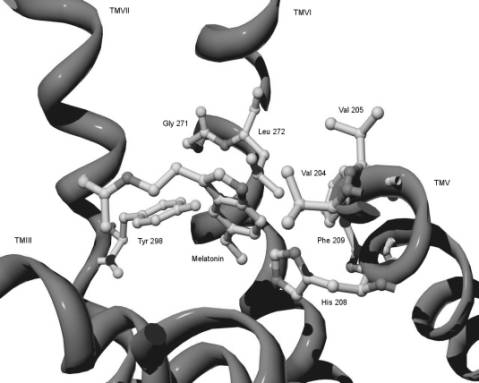
Fig. 3. The melatonin binding
site of melatonin receptor. The residues important for melatonin binding
(reported above) and melatonin docked into the binding site are shown.
According to our
model, Val 204 in TM V and Leu 272 in TM VI both occupy the area surrounding
the indole ring of the melatonin molecule (Fig. 3). Their physical properties
and proposed location indicate that they play a role in hydrophobic
interactions with the indole core of the ligand. This could be crucial for
adopting the correct orientation and/or stabilizing the melatonin molecule in
its binding pocket. The importance of Gly 271 to high-affinity melatonin
binding was previously reported for the MT I melatonin receptor [13]. We assume
that the effect of this mutation introducing bulkier and slightly polar Thr instead
of small and conformationally flexible Gly can be realized through the
affection of proper orientation of adjacent Leu 272 in the binding pocket. Val
205 is relatively aside from a docked ligand and thus it should not affect
binding parameters of the receptor. His 208 in TMV is proposed to participate
in a specific interaction with the 5-methoxy group of melatonin. The role of
His 208 in specific binding to the both subtypes of both mammalian melatonin
receptors was subsequently confirmed in experiments based on site-directed
mutagenesis showing the substantial increase of Kd value of the
mutant receptor [14]. According to our model Phe 209 does not have any specific
interaction with the melatonin molecule. In concord with our model we propose
that the hydroxy group of Tyr 298 through hydrogen bonding specifically
interacts with the 5-methoxy group in the melatonin molecule providing.
This research was supported by the Grant
Agency of the Czech Republic (Grant No. 309/02/1479), Internal Grant Agency of Academy
of Sciences (Grants No. A5011103 and A5011408) and by the Academy of Sciences
of the Czech Republic (Research Project No. AVOZ 501 1922). ZS, DS and RE
acknowledge support from the Ministry of Education of the Czech Republic (grant
no. LN00A141).
References
1. J. Vanecek, Physiol Rev, 78
(1998) 687-721.
2. M.L. Dubocovich et al., Front
Biosci, 8 (2003) 1093-1108.
3. K. Palczewski et al., Science,
289 (2000) 739-745.
4. N. Guex, M.C. Peitsch, Elektophoresis
18 (1997) 2714.
5. J.D.
Thompson et al., Nucl Acids Res 25 (1997) 4876-4884.
6. A. Sali, J.P. Overington, Protein Sci 3 (1994) 1582-1596.
7. B.R.
Brooks et al., J Comput Chem 4 (1983) 187-217.
8. R.A. Laskowski et al., J Appl
Crystallogr 26 (1993) 283-291.
9. D.A.
Pearlman et al., Computer Physics
Communications 91 (1995)
1-41.
10. C.
Chothia, A.M. Lesk, EMBO J 5 (1986) 823-826.
11. C. Venclovas et al.,
Proteins: Struct Funct Genet Suppl 1 (1997) 7-14.
12. V. Vlachova et al., Journal of Neuroscience 23
(2003) 1340-1350.
13. A.K.Gubitz, S.M. Reppert, Endocrinology 141(2000) 1236-1244.
14. S. Conway et al., Biochem.
Biophys. Res. Commun. 239 (1997) 418-423.