Current Challenges of Obtaining Crystals for X-ray and
Neutron Macromolecular Crystallography
M. Budayova-Spano, N. Junius, C. Berzin, Y. Sallaz-Damaz, J.L. Ferrer
Institut de Biologie Structurale, UMR5075 CEA-CNRS-UGA, Grenoble, FRANCE
monika.spano@ibs.fr
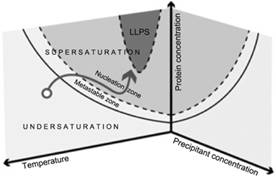
Knowledge of the phase diagram has key importance when designing and controlling a crystallization process for a substance. In the case of proteins, accurate phase diagram data is limited due to the complexity of their structure caused by the diversity of the amino acid residue groups that form proteins, with this process being easily influenced by environmental conditions. The solubility of a protein depends strongly on the protein−protein interactions as well as on the protein−solvent interactions. Any slight modification of the composition can influence the solubility dramatically, or even alter the nature of these macromolecules. Independently of the complexity of protein behaviour, the phase transformation is still governed by both the thermodynamics and the kinetics of the system. It is still possible to describe all this information in phase diagrams. In the case that crystallization conditions or nucleation points are identified the information can be plotted in phase diagrams, represented in a simplified form in Figure 1, and in this case the information that is provided relates to both thermodynamics and kinetics. Thermodynamic data are the solubility curves, the presence of metastable phases, polymorphs, liquid-liquid separation… They depend on multiple parameters such as temperature, pH, solvent, impurities, etc. In addition, kinetic trajectories in the phase diagram are relevant to control most of the final properties of the synthesized crystals. The path followed in the diagram controls the nucleation and growth of the crystals, and thus their number, size, and morphology.
Two new and emerging uses result in specific challenges for crystallization of proteins. In both, precise control of crystal size is essential. New approaches to serial (time-resolved) crystallography, where crystals in the 1-20 μm size are used to solve structures including those/structures of short-lived intermediates with reactions initiated by light or rapid mixing. Serial crystallographic methods are being increasingly used at synchrotron sources (serial synchrotron crystallography) due to advances in micro- and nano-focus beamlines, as well as at rapidly developing ultra-bright free-electron laser sources (serial femtosecond crystallography [1]), enabling structure determination of previously intractable proteins. At the other extreme are the requirements of the next-generation flagship neutron sources, such as the ESS (European Spallation Source, Lund). Because neutrons interact very weakly with matter, much larger, and ideally cubic crystals are needed with volumes of > 0.01 mm3 (i.e. 200 μm on a side) for neutron crystallography [2]. This is, however, the only way to visualise all of the protons in a protein structure, important information for drug design.
The detailed knowledge of the phase diagram is at the basis of the devices [3-6] we have developed especially with the focus on X-ray and Neutron Macromolecular Crystallography. The 1st generation instrument combines the use of temperature control and seeding and allows for grow of large crystals in crystallization batch [3]. A crystallization batch in the metastable zone is seeded with small protein crystals. The seeds are maintained inside this region of the phase diagram for as long as possible by doing a temperature step each time the crystal solution equilibrium is achieved. The temperature steps are repeated until crystals of suitable size for diffraction measurement are obtained.
The 2nd generation instrument, represented in Figure 2, adds new functionality to the first instrument thanks to a fluidic cell enabling to perform a temperature controlled dialysis crystallization experiment [4,5]. The new crystal growth apparatus combines accurate temperature control with control of the chemical composition of the crystallization solution and therefore it allows very sophisticated experiments to be performed. Systematic phase diagrams in multi-dimensional space can be investigated using far less protein material than previously. We have demonstrated that it can be beneficial to provide sufficient scattering volumes for neutron studies that require large-volume well-ordered single crystals. Based on this macro-scale instrument we have also conceived a miniaturizing apparatus that allows precise control of the experiment parameters using microfluidics [6]. The functional microfluidic chips integrating microdialysis with the volume less than 1 µL have been already successfully tested with model proteins. The microchips have multiple designs in order to achieve single or multiple crystallization experiments at the same time. They are transparent to X-ray radiation and allow performing in situ X-ray crystallography experiments at room temperature. The recently developed fluidic devices, once adapted, are expected to be useful in monitoring and controlling the crystallization processes of challenging biological macromolecules, such as membrane proteins.
|
|
|
|
Figure 1. Schematic view of a multidimensional phase diagram. The arrow illustrates a specific pathway taken during crystallization. |
Figure 2. Simplified view of a crystallization apparatus for temperature controlled flow cell dialysis with real time visualization. |

1. H.N. Chapman, Synchrotron Radiation News, 28, (2015), 20.
2. M.P. Blakeley, S.S. Hasnain, S.V. Antonyuk, IUCrJ, 2, (2015), 464.
3. M. Budayova-Spano, F. Dauvergne, M. Audiffren, T. Bactivelane, S. Cusack, Acta Cryst. D., D63, (2007), 339.
4. M. Budayova-Spano, Patent FR10/57354, UJF, (extension : EP117730945, US13821053, JP2013528746), (2010).