M. Šlouf
Ústav makromolekulární chemie AV ČR, Heyrovského nám. 2, 16206 Praha 6, Česká republika, e-mail: slouf@imc.cas.cz
Standardní rentgenostrukturní analýza monokrystalů je založena na tzv. modelu nezávislých atomů (= Independent Atom Model, IAM). V rámci IAM se předpokládá, že krystal se skládá z neinteragujících atomů. Každý atom je popsán pomocí polohových souřadnic x,y,z a teplotních parametrů Uij. Typickým výsledkem IAM je kompletní informace o polohách a teplotních pohybech atomů v krystalu, jinými slovy krystalová struktura.
Ačkoli IAM model se ukázal být velmi dobrou aproximací, je možno jít za jeho rámec, provést přesnější difrakční experiment a získat dodatečné informace týkající se atomů v krystalu. Pomocí kappa upřesňování (= Kappa Refinement, KR) [1] lze získat zejména informace o atomových nábojích. Pomocí multipólového upřesňování (= Multipole Refinement, MR) [2] lze získat úplný popis elektronové hustoty v krystalu, což v praxi znamená, že na speciálních mapách elektronových hustot můžeme pozorovat například kovalentní vazby a volné elektronové páry. V tomto pojednání bude stručně ukázáno:
v čem spočívá standardní rentgenostrukturní analýza a jaká jsou její omezení
jak získat atomové náboje pomocí metody kappa upřesňování
jak zviditelnit vazby v molekulách pomocí metody multipólového upřesňování
jak charakterizovat meziatomové interakce pomocí čísel s využitím metod topologické analýzy [3]
jak provádět difrakční experimenty a následné zpracování dat, abychom mohli získat informace o atomových nábojích pomocí kappa upřesňování a informace o elektronových hustotách v krystalu pomocí multipólového upřesňování
|
|
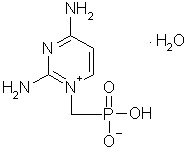
V dalším textu i se objeví praktické ukázky z práce, při které byly určeny atomové náboje a nábojové hustoty v látce s názvem monohydrát hydrogen[(2,4-diaminopyrimidin-1-io)methyl]fosfonátu [4, 5] (obr. 1), která bude dále označována jako H35 × H2O. Podrobnější informace o KR, MR a topologické analýze lze nalézt v původních zahraničních pracích [1, 2, 3] a ve dvou anglických učebnicích z nedávné doby [6, 7]. Stav problematiky na přelomu tisíciletí je shrnut ve velmi hezkém souborném článku [8]. V české literatuře jsou k dispozici dva přehledné referáty. První z nich [9] pojednává zejména o starších metodách X-X upřesňování a X-N upřesňování, které zde nebudou zmiňovány. Druhý [10] se zabývá především metodami KR a MR. |
| Obrázek 1. Elektronový strukturní vzorec molekuly H35·H2O |
Ve standardní rentgenostrukturní analýze se předpokládá, že krystal se skládá z navzájem neinteragujících atomů. Lze si jej tedy představit jako soubor sféricky symetrických atomů s pevně danou velikostí a pevně daným počtem elektronů. Odpovídající model se jmenuje model nezávislých atomů (Independent Atom Model = IAM) a elektronová hustota na atomu je definována následovně:
|
|
 |
Pomocí IAM modelu byly určeny všechny známé krystalové struktury, což dokazuje, že se jedná o opravdu velmi dobrou aproximaci. Tato skutečnost se dá vysvětlit například srovnáním hodnot elektronových hustot
[11]: maxima elektronové hustoty pro atomy z první řádky periodické soustavy se pohybují kolem 1000 eÅ-3, zatímco změny elektronové hustoty v důsledku vazby atomů se pohybují okolo 1 eÅ-3, což je číslo o tři řády menší. IAM model tedy správně popisuje drtivou většinu elektronové hustoty v krystalu, která je sféricky rozmístěna v okolí atomových jader. Každý atom je považován za „kuličku“, kterou charakterizujeme pomocí její polohy
(x, y, z) a jejích vibrací v krystalu (Uij nebo
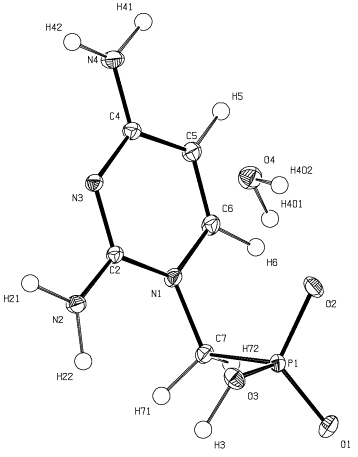
Uiso). Typickým výsledkem IAM modelu je krystalová struktura (obr. 2).
Aproximace použitá v IAM modelu funguje tím hůře, čím méně má atom elektronů. Krajním případem je atom vodíku s jediným elektronem. Pokud je vodík vázán kovalentní vazbou X-H (kde X = C, N, O), je jeho jediný elektron posunut směrem k atomu X. IAM model předpokládá, že elektronová hustota na atomu je sféricky symetrická a v maximu elektronové hustoty se nachází jádro atomu. Jelikož maximum elektronové hustoty pro atom vodíku je posunuto směrem k atomu X, při použití IAM nalezneme vodík v krystalové struktuře na nesprávném místě a výsledkem je efekt známý jako zdánlivé zkrácení X-H vazeb [12], které se může pohybovat řádově až v desetinách Å. Jiným efektem spojeným s nesférickým rozložením elektronové hustoty kolem atomů jsou asférické posuny. Efekt je poměrně jemný, ale stává se dobře patrným při porovnání rentgenografických a neutronografických dat. Neutrony difraktují na atomových jádrech a tudíž neutronová difrakce poskytuje přesné polohy jader atomů. Rentgenovy paprsky difraktují na elektronech, přičemž se v rámci IAM předpokládá, že v maximu elektronové hustoty se nachází jádro atomu, což však nemusí být vždy přesně splněno. Například v molekule vody je maximum elektronové hustoty pro atom kyslíku posunuto ve směru volného elektronového páru [13]. Asférické posuny jsou definovány jako rozdíly mezi neutronografickými a rentgenografickými polohami atomu a činí řádově tisíciny až setiny Å. |
| Obrázek 2. ORTEP diagram ukazující symetricky nezávislou část H35×H2O. |
Z dalších efektů, které odhalují některé nedostatky zjednodušeného modelu IAM, jmenujme ještě existenci dipólových momentů u molekul v krystalech a výskyt tzv. zakázaných difrakcí například u krystalů diamantu [14] nebo křemíku [15].
Kappa upřesňování (= Kappa Refinement, KR) je upřesňování krystalové struktury metodou nejmenších čtverců na základě kappa formalismu [1], což je poměrně jednoduchý model, který umožňuje popsat přesun elektronové hustoty mezi atomy díky tomu, že odděluje difrakci na vnitřních a valenčních elektronech atomu. V rámci modelu jsou atomy v krystalu stále považovány za sféricky symetrické, ale mohou si navzájem vyměňovat elektrony a mohou měnit svoji velikost. Elektronová hustota atomu je popsána vztahem:
|
|
kde funkce rcore je sféricky zprůměrovaná elektronová hustota příslušející vnitřním elektronům a rvalence je sféricky zprůměrovaná elektronová hustota příslušející valenčním elektronům atomu. Populační parametr valenční slupky Pv udává počet valenčních elektronů; atomové náboje je možno vypočíst jako (Nv-Pv), kde Nv je počet valenčních elektronů v izolovaném atomu. Radiální parametr k popisuje roztažení či smrštění valenční sféry atomu. Pokud platí k > 1, stejná elektronová hustota jako u izolovaného atomu se nachází v menší vzdálenosti r od jádra a tudíž je atom smrštěn ve srovnání s izolovaným atomem. Analogicky pro k < 1 je atom roztažen, protože stejná elektronová hustota se nachází ve větší vzdálenosti r od jádra. Funkce rcore a rvalence jsou pro každý atom pevně definovány. Parametry Pv a k se upřesňují spolu s polohovými a teplotními parametry atomů metodou nejmenších čtverců. Typickým výsledkem kappa upřesňování jsou právě hodnoty parametrů Pv a k, které poskytují informace o atomových nábojích a roztažení či smrštění atomů v krystalu (tab. 1).
| Tabulka 1. Výsledek KR pro sloučeninu H35·H2O. Atomové náboje
q jsou vypočítány jako Nv - Pv, kde
Nv je počet valenčních elektronů izolovaného atomu a
Pv je populační parametr valenční slupky. Jsou uvedeny pouze parametry pro nevodíkové atomy. |
|||
|
Atom |
Pv |
k |
q [e] |
|
P1 |
4.20(12) |
1.032(15) |
+0.80(12) |
|
O1 |
6.91(5) |
0.944(5) |
-0.91(5) |
|
O2 |
6.76(5) |
0.963(6) |
-0.76(5) |
|
O3 |
6.40(5) |
0.984(6) |
-0.40(5) |
|
O4 |
6.42(7) |
0.978(7) |
-0.42(5) |
|
N1 |
5.16(5) |
1.047(8) |
-0.16(5) |
|
C2 |
3.41(5) |
1.142(12) |
+0.59(5) |
|
N2 |
5.37(7) |
1.017(8) |
-0.37(7) |
|
N3 |
5.60(6) |
1.002(7) |
-0.60(6) |
|
C4 |
3.59(6) |
1.123(12) |
+0.41(6) |
|
N4 |
5.19(7) |
1.037(8) |
-0.19(7) |
|
C5 |
4.27(6) |
1.053(9) |
-0.27(6) |
|
C6 |
3.76(6) |
1.085(11) |
+0.24(6) |
|
C7 |
4.10(9) |
1.032(10) |
-0.10(9) |
V tabulce 1 si můžeme povšimnout, že experimentálně určené atomové náboje souhlasí s obecným konceptem elektronegativity. Nejelektronegativnějšími atomy ve struktuře jsou kyslíky, vesměs nesoucí poměrně vysoké záporné náboje. Naopak elektropositivní atomy fosforu, uhlíku či vodíku jsou nabity kladně. Pozoruhodný je zvláště záporný náboj na atomu dusíku N1, který je v rozporu ze strukturním elektronovým vzorcem (srovnej tab. 1, obr. 1 a obr. 2). Tento rozpor plyne z omezené platnosti oktetového pravidla, pomocí něhož se elektronové strukturní vzorce konstruují. Existenci experimentálně nalezeného záporného náboje na atomu N1 potvrzují i kvantově chemické výpočty [4]. Přesnější diskusi atomových nábojů na aromatickém kruhu H35·H2O je možné nalézt v ref. [4].
Model IAM považuje atomy v krystalu za izolované a nenabité. Při KR předpokládáme, že atomy si mohou vyměňovat elektrony a měnit svoji velikost. Nicméně jak v IAM, tak v KR jsou atomy považovány za sféricky symetrické. K popisu veškeré elektronové hustoty v krystalu je nutno použít pokročilejších modelů. V uplynulých 30 letech si největší popularitu získaly takzvané asférické pseudoatomové modely. Termín asférické značí, že elektronová hustota na jednotlivých atomech už není považována za sféricky symetrickou a termín pseudoatomové značí, že celková elektronová hustota v krystalu je poskládána z takzvaných pseudoatomů, což jsou nesférické elektronové hustoty středované na jádrech jednotlivých atomů. Nejpopulárnější asférický pseudoatomový model je multipólový model, založený na multipólovém formalismu [2]. Multipólové upřesňování (= Multipole Refinement, MR) je upřesňování krystalové struktury metodou nejmenších čtverců na základě multipólového formalismu. Elektronová hustota atomu je popsána vztahem:
|
|
kde vektor r a souřadnice r, q,
j jsou dány pomocí lokálního souřadnicového systému definovaného uživatelem na každém atomu. Celková elektronová hustota na atomu se získá sečtením tří členů. Význam prvních dvou členů je stejný jako v případě kappa upřesňování. Třetí člen, obsahující dvě sumace, mění atomovou elektronovou hustotu na nesférickou.
Rl jsou radiální funkce Slaterova typu a dlm± jsou angulární funkce, nazývané též multipólové hustotní funkce nebo jen multipólové funkce či multipóly. Tvary radiálních a angulárních funkcí jsou pevně dány. Angulární funkce se tvarem velmi podobají atomovým orbitalům a jejich orientace v prostoru je definována lokálním souřadnicovým systémem na daném atomu.
Plm± jsou multipólové populační parametry, udávající přesuny elektronů v rámci multipólových funkcí. Parametr
k’ popisuje roztažení či smrštění nesférické elektronové hustoty popsané pomocí radiálních a angulárních funkcí.
Při multipólovém upřesňování se kromě polohových a teplotních parametrů atomů upřesňují metodou nejmenších čtverců ještě parametry
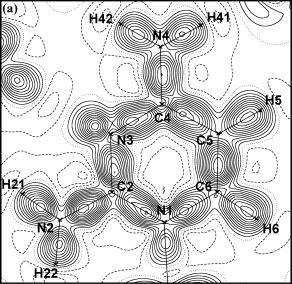
Pv, k, Plm± a k’. Prostřednictvím těchto parametrů je popsána nábojová hustota v krystalu, čehož lze využít pro vykreslování modelových map deformačních elektronových hustot (obr. 3a, 3b). Modelové mapy jsou typickým výsledkem multipólového upřesňování. Název
modelové vyplývá ze skutečnosti, že jsou založeny výhradně na parametrech získaných z multipólového modelu. Název
deformační říká, že mapy ukazují odchylky čili deformace od sféricky symetrické elektronové hustoty na atomech, tj. například kovalentní vazby nebo volné elektronové páry. Přesný popis modelových map deformačních elektronových hustot lze nalézt v literatuře
[2, 6].
 |
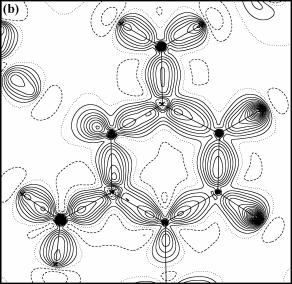 |
| Obrázek 3. Dynamická (a) a statická (b) mapa deformační elektronové hustoty v rovině aromatického kruhu H35·H2O. Kladné vrstevnice jsou plné, záporné čárkované a nulové tečkované čáry. Vrstevnice po 0.05 eÅ-3 (a) a 0.1 eÅ-3
(b). |
|
Máme-li kompletně popsánu elektronovou hustotu v krystalu, můžeme dále vypočítat celou řadu důležitých charakteristik molekul v krystalech. Lze počítat dipólové, kvadrupólové a vyšší elektrostatické momenty, elektrostatický potenciál v okolí molekuly, odhadovat popupace d-orbitalů v přechodných kovech a podobně [6-10].
Topologická analýza elektronové hustoty je, jak název napovídá, studium prostorového rozložení elektronové hustoty. Topologická analýza umožňuje kvantitativní zpracování informací obsažených na mapách elektronových hustot. Vstupem do topologické analýzy je funkce r(r), což je celková elektronová hustota v molekule nebo krystalu a výstupem jsou číselné údaje popisující veličiny jako je řád vazby, dvojný charakter vazby a podobně. Funkci r(r) můžeme získat buď z kvantově-chemických výpočtů nebo z multipólového upřesňování, což nabízí se velmi zajímavou možnost přímého srovnání teorie s experimentem. Z funkce r(r) můžeme vypočítat řadu dalších veličin, například:
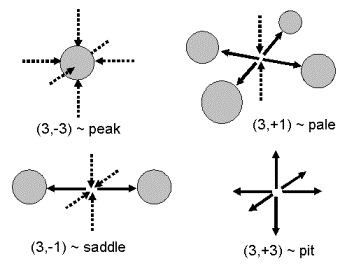 |
Problémem zůstává, jak vyextrahovat nějaké smysluplné údaje z tak ohromného množství dat: zatím jsme pouze z jedné funkce tří proměnných (r(r) = r(x,y,z) = r(x1, x2, x3)), získali několik dalších funkcí tří proměnných (dr(r)/dxi, d2r(r)/dxidxj, Ñr(r), Ñ2r(r) ). Naštěstí bylo ukázáno [3], že nejdůležitějšími prvky v třírozměrné elektronové hustotě jsou takzvané kritické body (= Critical Points = CP), kterých je v každé molekule či elementární buňce jen několik a nacházejí se v místech, kde jsou všechny první derivace elektronové hustoty nulové (dr(r)/dx = dr(r)/y = dr(r)/dz = 0). Kritické body se dále charakterizují pomocí druhých derivací elektronové hustoty (d2r(r)/dxidxj), přesněji řečeno pomocí křivostí l1, l2, l3: každý kritický bod je označen dvěma čísly, z nichž první udává počet nenulových křivostí (v případě třírozměrné elektronové hustoty je toto číslo vždy 3) a druhé udává součet znamének křivostí (zde jsou čtyři možnosti, viz obr. 4). Každé mapě elektronové hustoty dominují maxima elektronové hustoty v místech, kde se nacházejí atomy, což jsou podle topologické analýzy kritické body typu (3,-3). Z chemického hlediska jsou zpravidla nejzajímavější sedlové body vazeb, což jsou z hlediska topologické analýzy kritické body (3,-1), tzv. kritické body vazby (= Bond Critical Points = BCP). Pomocí hodnot výše definovaných funkcí v místě BCP lze každou vazbu kvantitativně popsat. Například řád vazby lze odvodit z hodnoty r(r) v BCP, dvojný charakter vazby lze získat pomocí hodnoty veličiny zvané elipticita (= elipticity: e = l1/l2 - 1) v BCP, kovalentnost vazby lze kvantitativně posoudit na základě hodnoty laplaciánu Ñ2r(r) v BCP a podobně. Podrobnější popis topologické analýzy lze nalézt v literatuře [3, 6, 8], příklad topologické analýzy aplikované na krystal H35·H2O lze nalézt v ref. [5]. |
| Obrázek 4. Všechny čtyři typy kritických bodů, které se mohou vyskytnout na třírozměrných mapách elektronové hustoty
r(r). Pomocí šedých koleček jsou schematicky znázorněny atomy. Pomocí šipek jsou schematicky znázorněny nárůst (přerušované šipky) a pokles (plné šipky) elektronové hustoty. Kritické body jsou označeny čísly a anglickými názvy. Je vidět, že kritickým bodům (3,-3) odpovídají atomy, bodům (3,-1) sedlové body vazeb, bodům (3,+1) středy kruhů a konečně bodům (3,+3) odpovídají lokální minima na mapách elektronových hustot. |
Na možnost studovat meziatomové interakce pomocí rentgenostrukturní analýzy upozornil P. Debye již roku 1915, nicméně přesnost měření difrakcí nebyla zdaleka postačující, takže k prvním experimentům došlo až v 60. a 70. letech minulého století. Metody KR a MR byly popsány na samém konci 70. let [1, 2]. V 80. a 90. letech obor postupně vyzrál, na konci 90. let už se objevily i první učebnice [5, 6]. V České republice se této tématice zřejmě nikdo nevěnoval a první studie se tak objevily až ve třetím tisíciletí [4].
Pro určování atomových nábojů pomocí kappa upřesňování je nutné mít k dispozici přesnější a kvalitnější data než je obvyklé ve standardní rentgenostrukturní analýze. Ještě kvalitnějších dat je zapotřebí pro určování nábojových hustot v krystalu pomocí multipólového upřesňování. Data se obvykle měří za nízkých teplot kolem 100 K, aby se minimalizoval teplotní pohyb atomů, který při studiích nábojových hustot působí rušivě. Dále je nutné změřit difrakce s vysokými difrakčními úhly q, aby bylo dosaženo vyššího rozlišení na mapách elektronových hustot. Pro standardní rentgenostrukturní analýzu postačují nízkoúhlové difrakce se sinq/l <= 0,6 Å-1, kde l je vlnová délka použitého rentgenového záření. Pro určování atomových nábojů je vhodné změřit i difrakce se sinq/l >= 0,6 Å-1 a pro studia nábojových hustot jsou nezbytné i vysokoúhlové difrakce se sinq/l >= 0,9 Å-1.
Difrakční data je možno měřit buď s použitím bodových nebo plošných detektorů. Praxe ukázala, že bodové detektory jsou zpravidla přesnější pokud jde o měření jednotlivých difrakcí, ale měření objemů dat nutných pro metodu MR trvá týdny až měsíce pro jeden krystal. U plošných detektorů je měření značně rychlejší, trvá zpravidla jen několik hodin (pro metodu KR) nebo dní (MR); v přesnosti se nakonec plošné detektory minimálně vyrovnají bodovým, pokud jsou data měřena s velkou redundancí. Používá se redundancí kolem 5 i více, přičemž redundance 5 znamená, že jedna reflexe je změřena v průměru 5x.
Pokud jde o studované krystaly, byly zpočátku studovány především dobře difraktující krystaly (větší přesnost měření silných reflexí), obsahující jen několik málo atomů v symetricky nezávislé části (větší rychlost výpočtů), krystalizující v centrosymetrických prostorových grupách (přesnější určení fází strukturních faktorů), sestávající z lehkých prvků z první a druhé řady periodické tabulky (příznivý poměr vnitřních a valenčních elektronů - vazebné efekty se týkají prakticky výhradně valenčních elektronů a jsou tudíž snáze pozorovatelné na lehkých atomech). S vývojem přístrojové i výpočetní techniky se začínaly studovat i větší látky (velké organické molekuly) krystalizující v necentrosymetrických grupách (oligopeptidy) a obsahující těžší prvky (do první řady přechodných kovů včetně).
Jak už bylo diskutováno výše, jsou změny celkové elektronové hustoty v krystalu, které vzniknou v důsledku mezivazebných velmi jemné. Aby bylo možno tyto odchylky vůbec odhalit, je nutno měřit difrakční data co nejpřesněji a pokud krystal obsahuje těžší prvky, jsou bezpodmínečně nutné přesné absorpční a extinkční korekce, které se ostatně doporučují i u krystalů obsahujících jen lehčí prvky.
Standardní rentgenostrukturní analýza monokrystalů založená na IAM postačuje pro naprostou většinu případů. Jsou-li k dispozici nadprůměrně kvalitní krystaly, je možno využít též metod KR a MR, čímž lze získat řadu dalších informací, závažných jak z teoretického, tak z praktického hlediska. Pokud jde o teorii, metody KR a MR příspívají k pochopení vazeb v molekulách a umožňují experimentálně ověřovat kvantově mechanické výpočty. V praxi lze použít například experimentální atomové náboje při návrhu organických syntéz nebo elektrostatické potenciály, které hrají důležitou roli při molekulárním rozpoznávání.
Tato práce byla podporována prostřednictvím Grantové agentury České republiky (projekt AVOZ 4050913 a grant 203/99/MO037).