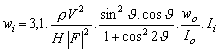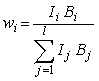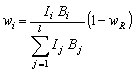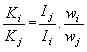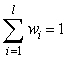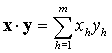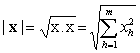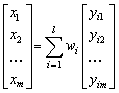![]()
(1)
Jaroslav Fiala
Nové technologie – Výzkumné centrum, Západočeská univerzita Plzeň,
Universitní 22, 306 14 Plzeň
O kvantitativní rtg. difrakční fázové analýze najdeme mnoho informací ve zvláštních kapitolách různých souborných publikací, z nichž můžeme zvlášť doporučit knihy
[1-3]. Avšak monografie, pojednávající výhradně o rtg. difrakční kvantitativní fázové analýze jsou na světě jenom tři
[4-6]. Z nich může sloužit jako mimořádný dobrý průvodce po oblasti rtg. difrakční kvantitativní fázové analýzy zejména poslední Zevinova kniha
[6]. U nás vyšly o této problematice dva souborné články v časopise Silikáty
[7, 8] a tamtéž se objevil i článek shrnující relevantní názvosloví
[9]. Kvantitativní fázové analýze je věnována také 10. kapitola knížky
[10], na jejímž vydání se podílela (česká a slovenská) Krystalografická společnost.
Podíl fáze v analyzované směsi je přímo úměrný intensitě difrakcí (difrakčních linií) té fáze. Intensita
Ii zvolené difrakční čáry i-té fáze
|
|
(1) |
kde
Pi … přístrojový faktor (mění se od přístroje k přístroji podle detailů experimentálního uspořádání);
Qi … faktor daný ideální krystalovou strukturou dané fáze (polohami atomů v základní buňce krystalové struktury i-té fáze a jejich teplotními kmity kolem těchto poloh);
Ti …. faktor daný reálnou strukturou difraktujícího preparátu, tj. tím, jak velké jsou krystalky jednotlivých fází obsažených v preparátu, jaký je jejich tvar, orientace, umístění a nejrůznější odchylky od ideální krystalové struktury;
wi …. hmotnostní koncentrace i-té fáze v preparátu.
Intensita zvolené difrakční linie dané (i-té) fáze nezáleží tedy jenom na tom kolik je té fáze ve vzorku (wi) a jaká je to fáze (a kterou její difrakční linii jsme si zvolili jako analytickou) – Qi, ale také na tom, jaké se použilo experimentální uspořádání. O tento přístrojový vliv (Pi) je třeba naměřené hodnoty intensit redukovat, abychom je mohli použít pro výpočet obsahu jednotlivých fází ve zkoumaném preparátu (pro kvantitativní fázovou analýzu toho preparátu). Vliv reálné struktury na intensitu difrakčních linií (Ti) se často (v první aproximaci) zanedbává. Ve skutečnosti je to však nejdůležitější zdroj chyb (příčina neurčitosti výsledků) kvantitativní fázové analýzy a proto bude tento vliv podrobně diskutován ve zvláštní kapitole.
Chceme-li z naměřených hodnot intensit difrakčních linií Ii vypočítat hmotnostní koncentrace wi jednotlivých fází přítomných v analyzovaném preparátu
|
|
musíme intensity difrakčních linií Ii redukovat o vliv experimentálního uspořádání
Pi a vzít v úvahu o jakou se jedná fázi (a jakou difrakční linii jsme si vybrali pro tu fázi jakožto analytickou) –
Qi. Tomu se říká kalibrace. Kalibrace se provádí různými způsoby, většinou „metodou vnějšího standardu“ nebo „metodou vnitřního standardu“. Teorie difrakce umožňuje kalibraci „obejít“ výpočtem faktorů
Pi a Qi na základě známé struktury určované fáze při zvoleném experimentálním uspořádání. V tom případě se někdy hovoří o „absolutní“ kvantitativní fázové analýze nebo o kalibračních konstantách získaných výpočtem („calculated patterns“).
V tomto případě měříme nejen intensity Ii zvolených linií určovaných l fází (i = 1, 2, ….., l) analyzovaného vzorku, ale navíc ještě intensitu Io referenční linie nějakého dalšího vzorku, který jsme si vybrali jako standard („vnější standard“). Volba vnějšího standardu a jeho referenční linie není v podstatě ničím omezena; zejména nemusí mít žádnou souvislost se složením a strukturou analyzovaného vzorku. Kromě toho musíme změřit hmotnostní součinitel zeslabení mm analyzovaného vzorku. Pro obsah (hmotnostní koncentraci) wi i-té fáze v analyzovaném vzorku platí
|
|
(2) |
kde Ci je kalibrační konstanta pro i-tou fázi (zvolenou linii) a použité experimentální uspořádání.
Kalibrační konstanty Ci; i = 1, 2, …., l pro l fází, jež nás zajímají, můžeme určit ze vzorce (2), pokud máme k dispozici preparáty známého fázového složení. Nemáme-li takové preparáty k dispozici, seženeme si nějaké vzorky (jejichž kvantitativní fázové složení neznáme) o počtu n, které uvažovaných l fází obsahují, a to tak, aby n >= k, kde k >= l je celkový počet různých fází vyskytujících se v těch vzorcích. Potom pro každý z těch n vzorků platí
|
|
a tedy
|
|
kde Ii resp. Ci (i = 1, 2, …, k) jsou intensity zvolených analytických linií resp. kalibrační konstanty těch k fází přítomných ve vzorcích, mm je hmotnostní součinitel zeslabení uvažovaného vzorku a Io je intensita referenční linie vnějšího standardu.
Změříme-li pro každý (j-tý; j = 1, 2, …, n) z těch vzorků jeho hmotnostní součinitel zeslabení
mm1, mm2, …,
mmn a intensitu Iji zvolené analytické linie každé
(i-té; i = 1, 2, …, k) fáze, dostaneme soustavu n rovnic.
|
|
(3) |
a jejím řešením hledané kalibrační konstanty C1, C2, …, Cl (l <= k). V případě, že máme k dispozici čisté preparáty jednotlivých fází, bude n = k = l a matice soustavy (3) bude diagonální, takže pro kalibrační konstanty dostaneme ze (3) vzorce
|
|
V tomto případě musíme ke každému analyzovanému vzorku přimíchat určitý (hmotnostní) podíl wo nějaké další látky, kterou jsme si vybrali jako standard („vnitřní standard“). Volba vnitřního standardu není v podstatě ničím omezená. Omezení je však v tom, že vnitřní standard musíme k analyzovanému vzorku „přimíchat“; analyzovaný vzorek musí být tedy (snadno a beze změny fázového složení) dělitelný: pro kompaktní a těžko nebo problematicky dělitelné vzorky se kalibrační metoda vnitřního standardu nehodí. Na difraktogramu takto modifikovaného analyzovaného vzorku, tj. analyzovaného vzorku, ke kterému je přidán vnitřní standard, pak měříme nejen intensity Ii zvolených linií určovaných l fází (i = 1, 2, …, l), ale navíc ještě intensitu Io referenční linie přimíchaného vnitřního standardu. Pro obsah (hmotnostní koncentraci) wi i-té fáze v analyzovaném vzorku platí
|
|
(4) |
kde Di je kalibrační konstanta pro i-tou fázi (zvolenou linii) a použité experimentální uspořádání.
Kalibrační konstanty Di; i = 1, 2, …, l pro l fází jež nás zajímají můžeme určit ze vzorce (4) pokud máme k dispozici preparáty známého fázového složení. Nemáme-li takové preparáty k dispozici, seženeme si nějaké vzorky (jejichž kvantitativní fázové složení neznáme) o počtu n, které uvažovaných l fází obsahují, a to tak aby n >= k, kde k >= l je celkový počet různých fází vyskytujících se v těch vzorcích. Ke každému tomu vzorku přidáme hmotnostní podíl wo zvoleného vnitřního standardu. Potom pro každý z těch n vzorků platí
|
|
a tedy
|
|
kde Ii resp. Di (i = 1, 2, …, k) jsou intensity zvolených analytických linií resp. kalibrační konstanty těch k fází přítomných ve vzorcích a Io je intensita referenční linie přimíchaného vnitřního standardu.
Změříme-li pro každý (j-tý; j = 1, 2, …, n) z těch n vzorků intensitu Iji zvolené analytické linie každé (i-té; i = 0, 1, 2, …, k) z k+1 fází přítomných ve vzorcích (jako nultou fázi označujeme vnitřní standard) dostaneme soustavu n rovnic.
|
|
(5) |
a jejím řešením hledané kalibrační konstanty D1,
D2, …, Dl, (l <= k). V případě, že máme k dispozici čisté preparáty jednotlivých fází, bude
n = k = l a matice soustavy (5) bude diagonální, takže pro kalibrační konstanty dostaneme z (5) vzorce
|
|
Teorie difrakce rentgenového záření je vypracována tak, že umožňuje vypočítat přístrojový faktor Pi a faktor Qi daný (ideální) krystalovou strukturou ve vzorci (1), které potřebujeme pro výpočet hmotnostní koncentrace wi
|
|
z intensity Ii zvolené difrakční čáry [11-16, 2]. Tak pro Braggovo-Brentanovo parafokusační uspořádání (pro „klasický“ práškový difraktometr Parrishovy konstrukce [17-20]), nepolarisované záření CuKa a preparát o tloušťce větší než pětinásobek převrácené hodnoty lineárního součinitele zeslabení ve vzorku platí
|
|
(6) |
kde q je difrakční úhel zvolené (analytické) difrakční linie určované fáze,
H její faktor četnosti, F strukturní faktor, V objem základní buňky (jejíž rozměry jsou vyjádřeny v jednotkách
10-10m) krystalové struktury určované fáze, r její hustota (v jednotkách
gcm-3), Io intensita difrakční linie (113) korundu, který je v analyzovaném vzorku obsažen (například byl do něho přimíchán) ve hmotnostní koncentraci
wo%.
Pro jiná uspořádání nalezneme příslušné vzorce v tabulkách [21-23]. Hodnoty koeficientů (jež slouží jako kalibrační konstanty) ve vztahu
|
|
(7) |
vypočtené na způsob formule (6) při určitém experimentálním uspořádání byly publikovány pro řadu látek
[24-31]. Možnost vypočítat kalibrační konstanty na základě známé struktury určovaných látek je vítaná zejména v případě, že tyto látky nemáme momentálně fyzicky k dispozici. I v tomto případě musel ovšem už dříve někdo difraktogram té látky (jejího monokrystalu) změřit, aby její strukturu určil.
Ať už při kvantitativní rtg. difrakční fázové analýze používáme kalibrační konstanty
Ci pro metodu vnějšího standardu (2) nebo kalibrační konstanty
Di pro metodu vnitřního standardu (4) či kalibrační konstanty
Ei vypočtené na základě známé struktury určovaných látek (7), platí pro poměry hmotnostních koncentrací
wi/wj i-té a j-té fáze (látky) přítomné v analyzované směsi analogické vztahy
|
|
(kde Ii a Ij jsou intensity difrakční linie i-té a j-té fáze, jež jsme si vybrali pro provádění kvantitativní fázové analýzy). To znamená, že poměr hmotnostních koncentrací dvou fází wi/wj je úměrný poměru intensit jejich analytických difrakčních linií Ii/Ij a koeficient této úměrnosti neobsahuje už žádné jiné veličiny než příslušné intensitní faktory (kalibrační konstanty). Označíme-li je obecně jako Bi (Bi = Ci pro metodu vnějšího standardu, Bi = Di pro metodu vnitřního standardu a Bi = Ei pro případ, že kalibrační koeficienty určujeme výpočtem na základě známé struktury příslušných fází), pak tedy
|
|
(8) |
To umožňuje kalibrační konstanty unifikovat: Zvoleným jednotným způsobem (pomocí vnějšího standardu, pomocí vnitřního standardu nebo výpočtem na základě známé struktury) určíme předem a jednou provždy kalibrační konstanty B1, B2, …, Bl, všech fází, které se budou vyskytovat v analyzovaných vzorcích. A při vlastním rozboru zkoumaného vzorku, který ovšem musí být proveden tím (jednotným) způsobem a v tom experimentálním uspořádání pomocí něhož byly určeny kalibrační konstanty (intensitní faktory) Bi, pak už nepotřebujeme ani vnější ani vnitřní standard (někdy se mluví o „bezstandardové metodě“); pro hmotnostní podíl wi i-té fáze (i = 1, 2, …, l) v tom vzorku dostaneme pomocí vztahu (8) na základě toho, že
|
|
(9) |
vzorec
|
|
(10) |
Tento vzorec ovšem platí pouze tehdy, když se ve zkoumaném preparátu nevyskytují jiné fáze než těch l, pro něž jsme během přípravy analytického programu určili kalibrační konstanty B1, B2, …, Bl a při vlastní analýze změřili intensity I1, I2, …, Il vybraných difrakčních linií.
V případě, že se ve zkoumaném preparátu vyskytují ještě nějaké jiné fáze (např. amorfní složka) a jejich celkový hmotnostní podíl je
wR, platí místo (10) vztah
|
|
(11) |
V některých případech se při kvantitativní rtg. difrakční fázové analýze používají zvláštní postupy, „šité na míru“ pro určitý analytický program.
Sem patří například metoda referenčních směsí. V laboratoři se vyrobí (syntetisují) preparáty, jejichž složení vytváří řadu (síť, vícerozměrnou matici), (více nebo méně) hustě pokrývající obor složení vzorků, jež do laboratoře přicházejí v rámci určitého analytického programu. Z těchto vzorků (známého složení) se pořídí referenční difraktogramy. A ty se pak porovnávají s difraktogramem neznámého vzorku, jemuž se na základě toho přisoudí složení odpovídající té referenční směsi, jejíž difraktogram je difraktogramu analyzovaného vzorku nejvíce podobný. Aby to fungovalo, musí se samozřejmě používat stále jedno a totéž experimentální uspořádání.
Jindy se používá metoda homologických párů. To jsou páry difrakčních linií dvou fází, které jsou v určitém poměru. Když tedy zjistíme, že na difraktogramu analyzovaného vzorku jsou určité dvě čáry zkoumaných fází (přibližně) stejně intensivní, znamená to, že poměr jejich koncentrací v tom vzorku je (přibližně) rovný odpovídající hodnotě z tabulek homologických párů. I v tomto případě je při analýze neznámého vzorku nutno použít totéž uspořádání, které bylo použito při sestavování příslušné tabulky homologických párů. Předností této metody jest, že vychází z identifikace stejně intensivních (difrakčních) linií. Je to tedy svým způsobem metoda kompensační, jež eliminuje nelineárnost odezvy detektoru.
Dalším takovým postupem je metoda přídavku (doping method). Koncentraci
w určité fáze v analyzovaném vzorku stanovíme na základě změřené hodnoty intensity zvolené linie té fáze na difraktogramu analyzovaného vzorku
(I), na difraktogramu analyzovaného vzorku, ke kterému jsme přidali hmotnostní zlomek
w1 určované fáze (I1) a posléze na difraktogramu analyzovaného vzorku, ke kterému jsme přidali hmotnostní zlomek
w2 té fáze (I2) podle vzorce
|
|
Metoda přídavku je pracná, ale může být výhodná například v případě, že není k dispozici vhodný vnitřní standard. Nehodí se pro analýzy velkých sérií, ale může se vyplatit, když se jedná o přesnou analýzu malého počtu vzorků. Podrobný rozbor šíření chyb ukazuje, že metoda přídavku je efektivní pro analýzy minoritních fází, asi tak do 20% obsahu [6].
Pro střední a vyšší obsahy určované fáze se naopak může dobře uplatnit metoda zřeďovací (dilution method). Ta spočívá v tom, že analyzovaný vzorek ředíme materiálem, který může být amorfní a v případě, že je krystalický nemusí obsahovat žádnou z fází obsažených v analyzovaném vzorku. Změříme intensitu zvolené linie určované fáze na difraktogramu analyzovaného vzorku (I) a na difraktogramu zřeďěného vzorku, který vznikl tak, že jsme k analyzovanému vzorku přidali v% hmotnostních ředidla (I1). Pro (hmotnostní) koncentraci w určované fáze v analyzovaném vzorku pak platí
|
|
kde m resp. mv je hmotnostní součinitel zeslabení analyzovaného vzorku resp. ředidla a
Io je intensita zvolené linie určované fáze na difraktogramu čistého preparátu této fáze. Samozřejmě, že všechny tři difraktogramy musí být pořízeny při tomtéž experimentálním uspořádání. Analýza rozptylu ukazuje, že optimální poměr
I1/Io jest I1/Io = 1/3
[6]. Také zřeďovací metoda je pracná a její aplikace se vyplatí jenom v určitých zvláštních situacích, když univerzální techniky vnitřního či vnějšího standardu jsou z toho nebo jiného důvodu v daném konkrétním případě málo účinné.
Nejdůležitějším zdrojem chyb (příčinou neurčitosti) kvantitativní rtg. difrakční fázové analýzy je reálná struktura. Ta ovlivňuje intensitu difraktovaného záření a tento vliv není jednoduché (a někdy dokonce ani možné) od vlivu, který má na intensitu difrakcí fázové složení, odlišit. Reálnou strukturou rozumíme velikost (velikostní distribuci) krystalků, jejich tvar (tvarovou distribuci), prostorové a směrové rozložení a rozličné strukturní defekty (odchylky od ideální krystalové struktury). Tak už jenom proto, že krystalky jsou velké, dochází k rozptylu naměřených hodnot intensity difraktovaného záření, což může přesnost výsledků kvantitativní fázové analýzy podstatně snížit. Jsou-li krystalky velké asi 40
mm, bude to představovat zhoršení o 66%, u krystalků velkých 10
mm o 8% a dokonce i v případě, že krystalky jsou velké jenom 1
mm, zhorší to neurčitost výsledků kvantitativní fázové analýzy o nezanedbatelné 1%
[32]. Hrubozrnnost zmenšuje přesnost kvantitativní fázové analýzy tím více, čím menší je podíl určované fáze ve vzorku. Jak velký může být vliv reálné struktury na intensitu difrakčních linií a k jak velké chybě může v důsledku toho dojít při kvantitativní fázové analýze, to ilustrují údaje publikované v práci
[33]: pro čtyři různé preparáty kalcitu a difrakční linii (014) změřili autoři této práce takovéto hodnoty kalibračních faktorů: 4,2; 2,0; 2,2 a 4,0 (jestliže by reálná struktura neměla na difrakci vliv, musela by tato čtyři čísla být stejná). Pro dva různé preparáty pyritu a difrakční linii (311) byly naměřeny hodnoty kalibračních faktorů 0,7 a 1,5 a pro tři různé preparáty křemene a difrakční linii (101) byly změřené hodnoty kalibračních faktorů 2,7; 5,1 a 4,5. Do kalibračních faktorů příslušných fází
Ki, jež vyjadřují vztah
|
|
mezi jejich koncentrací wi resp. wj ve zkoumaném vzorku a intensitou Ii resp. Ij jejich analytické linie, nepřispívají totiž jen kalibrační konstanty Bi resp. Bj z rovnice (8), která platí pouze za předpokladu, že vliv reálné struktury lze zanedbat, ale i faktory reálné struktury Ti resp. Tj z rovnice (1):
To, že vliv reálné struktury není možné zanedbat znamená, že faktory reálné struktury Ti se mění od vzorku k vzorku. Faktory Ki v tom případě nejsou konstanty a pro kalibraci při kvantitativní fázové analýze ztrácejí význam.
Nejčastěji používaným opatřením pro vyloučení (nebo alespoň potlačení) vlivu reálné struktury je preparace, kterou se snažíme krystalky analyzovaného vzorku rozmělnit do velikosti kolem jednoho milimetru a dosáhnout toho, aby jejich orientace byla zcela náhodná. Přesto, že k tomu účelu byla vyvinuta řada důmyslných postupů [5], nedaří se tímto způsobem vliv reálné struktury eliminovat ve všech případech a se stoprocentní účinností. U některých materiálů se z technických důvodů rozmělňování krystalků nedaří nebo je spojeno s nebezpečím, že při něm dojde k fázovým transformacím, jež složení preparovaného vzorku změní. Často při rozmělňování vzorku dochází k amorfisaci, čímž vznikne systematická chyba, pokud se při kvantitativní fázové analýze použijí unifikované kalibrační konstanty (neurčitost hodnoty wR ve vzorci (11). Ta chyba je tím větší, že amorfní fáze vzniká často jako obálka zrn, která oslabuje primární záření pronikající do jejich krystalického jádra, jakož i záření difraktované, které vychází z krystalického jádra zrn skrze jejich amorfní obálku. Vliv amorfní fáze disponované na obálkách zrn je (mnohem) větší než by odpovídalo obsahu amorfní fáze (kdyby byla v preparátu rozložena rovnoměrně).
Výsledkem toho je, že chyba (neurčitost výsledků) rtg. difrakční kvantitativní fázové analýzy je 1 – 10% absolutních (obsahu kterékoli fáze analyzovaného vzorku). Jediná cesta, která slibuje podstatně a plošně zvýšit přesnost určení obsahu jednotlivých fází ve zkoumaném vzorku, vede přes faktorovou analýzu [34], tj. bilineární modelování [35-38].
Z materiálu, jehož fázové složení máme kvantitativně určit, vyrobíme n frakcí (jednou „frakcí“ je původní dodaný vzorek). Tyto preparáty budou mít kvalitativně shodné, kvantitativně však odlišné fázové složení. Pokud se poštěstí, aby i reálná struktura takto připravených frakcí byla shodná, pak kalibrační faktory l fází jež nás zajímají Ki (i = 1, 2, …, l) budou konstantní a můžeme je určit metodou vnitřního standardu, tj. řešením soustavy rovnic (5), kde Iji je intensita zvolené analytické linie i-té fáze na difraktogramu j-té frakce (j = 1, 2, …, n), wo je hmotnostní podíl zvoleného vnitřního standardu a Ijo je intensita referenční linie vnitřního standardu, přimíchaného k j-té frakci. Hodnoty Ki vypočtené pro jednotlivé fáze i = 1, 2, …, l analyzovaného preparátu, které nás zajímají, z rovnic (5) formálně jako Di, budou ve skutečnosti zahrnovat i vliv reálné struktury Ti (identický pro všechny frakce separované z analyzovaného vzorku), takže pro jiný zkoumaný vzorek a tytéž fáze vyjdou (aplikací téže procedury) jiné hodnoty Ki (viz rovnici (13)) prostě proto, že ten vzorek má jinou reálnou strukturu. Pro každý vzorek, jehož rozbor tímto způsobem provedeme, budou však výsledky kvantitativní fázové analýzy na vliv reálné struktury korigovány. V případě, že zkoumaný vzorek nelze dělit a separovat na různé frakce, je možno ten postup použít v poněkud pozměněné podobě, metodou vnějšího standardu, když se nám podaří sehnat n-1 dalších vzorků (neznámého složení ale) stejné paragenese (tedy vzorky, o nichž se dá předpokládat, že budou mít tutéž reálnou strukturu) jako zkoumaný vzorek. Kalibrační faktory l fází Ki (i = 1, 2, …, l), které nás zajímají ve zkoumaném vzorku pak budou konstantní a můžeme je vypočítat řešením soustavy rovnic (3), kde Iji je intensita zvolené analytické linie i-té fáze na difraktogramu j-tého vzorku (j = 1, 2, …, n), mmj je hmotnostní součinitel zeslabení toho vzorku a Io je intensita referenční linie vnějšího standardu. Opět platí, že hodnoty Ki, vypočtené pro jednotlivé fáze zkoumaného vzorku, jež nás zajímají, z rovnic (3) formálně jako Ci, budou ve skutečnosti zahrnovat i vliv reálné struktury Ti. Pokud se nám podaří najít těch doprovodných n-1 vzorků tak, aby jejich reálná struktura byla stejná jako je reálná struktura zkoumaného (primárně zadaného) vzorku, pak hodnoty kalibračních faktorů Ki budou korektní. Poznáme to podle toho, že jejich neurčitost odhadnutá řešením soustavy (3) bude malá. Jestliže neurčitost vyjde velká, znamená to, že doprovodné vzorky, které jsme analyzovali spolu s primárně zadaným zkoumaným vzorkem, nemají stejnou reálnou strukturu a hodnoty kalibračních faktorů získaných tímto způsobem nejsou korektní. To, že se vedle hodnot kalibračních faktorů dozvíme i jsou-li správné čili nic, je velkou předností tohoto postupu kalibrace.
Velká naděje na zpřesnění kvantitativní rtg. difrakční fázové analýzy se vkládala do multivariantní kalibrace, tedy toho, že se pro výpočet fázového složení využije mnoho píků
[25, 39] a posléze celý „difrakční profil“ („full trace“ nebo „whole-profile pattern“, tj. směrová závislost intensity difraktovaného záření v širokém intervalu Braggových úhlů s krokem, který je mnohem menší než šířka píku)
[40]. Difraktogramy se přitom pojímají jako vektory (body) mnohorozměrného abstraktního „prostoru difrakčních obrazů“
[41-45] daného digitalizací difraktogramu, tak jak je registrován v několika tisících krocích („kanálech“). Ty kanály (směry v nichž je registrována intensita difraktovaného záření) po řadě očíslujeme 1, 2, 3, …,
m a intensity změřené v těchto směrech označíme x1,
x2, …, xm; difraktogramu pak přiřadíme m-tici (číselný vektor)
x = (x1, x2, …, xm). Takové přiřazení je isomorfismus v tom smyslu, že vektor difraktogramu směsi je lineární superposicí vektorů difraktogramů jejích jednotlivých komponent, přičemž koeficienty té lineární superposice se rovnají podílům (hmotnostním koncentracím) příslušných komponent v analyzované směsi. Provést kvantitativní fázovou analýzu dané směsi pomocí rtg. difrakce znamená vyjádřit její difraktogram x superposicí difraktogramů
y1 = (y11, y12, …, y1m),
y2 = (y21, y22, …, y2m), …,
yl = (yl1, yl2, …, ylm) jejích komponent, tedy nalézt čísla
w1, w2, …, wl, pro něž
|
|
a která co nejlépe splňují vztah
|
|
(14) |
t.j. minimalizují residuum
|
|
Z podmínky stacionárnosti
|
|
dostáváme pak normální soustavu rovnic
x
. y1 = w1y1.y1 + w2y2.y1
+ … + wlyl.y1
x
. y2 = w1y1.y2 + w2y2.y2
+ … + wlyl.y2
…………………………………………..
x . yl = w1y1.yl + w2y2.yl + … + wlyl.yl
pomocí které fázové složení určujeme. V těchto výrazech je symbolem
|
|
označen skalární součin vektorů takto definovaného euklidovského prostoru difraktogramů
x = (x1, x2, …, xm) a y = (y1, y2, …, ym);
euklidovská norma vektoru difraktogramu
|
|
Naděje na zlepšení přesnosti kvantitativní rtg. difrakční fázové analýzy multivariantní kalibrací vycházela z racionální úvahy o tom, že rovnice (14) v souřadnicové representaci
|
|
(15) |
obsahuje při m rovném několika tisícům (krokům, kanálům) velký objem redundantní informace. Objem mnohonásobně převyšující informaci, již získáváme při konvenčním (univariantním) postupu, když pro každou fázi změříme integrální intensitu jediné difrakční linie. Praxe (provádění kvantitativní rtg. difrakční fázové analýzy Rietveldovou metodou) však ukázala, že naděje vkládané v tuto metodu se nenaplnily. Příčinou je to, že reálná struktura ovlivňuje difraktogramy yi jednotlivých komponent analyzované směsi yi = yi (Ti) = {yi1 (Ti1), yi2 (Ti2), …, yim (Tim)} tak, že do soustavy (15) vnese dalších (obecně až) m x l neurčitých parametrů Tij (i = 1, 2, …, l; j = 1, 2, …, m), což informační zisk z proměření difraktogramů ve velkém počtu (m) kanálů znehodnotí. Ani s pomocí Rietveldovy metody nejsme tedy zpravidla schopni určit obsah jednotlivých komponent zkoumané směsi přesněji než na několik (1-10) procent absolutních, pokud nepoužijeme faktorovou analýzu.
Pro rtg. difrakční techniku kvantitativní fázové analýzy je specifické, že vypovídá o objemu materiálu, který zasahuje do hloubky několika mikrometrů pod povrch ozařovaný primárním rentgenovým svazkem (na rozdíl od světelné mikroskopie pracující v uspořádání na odraz, jež postihuje pouze to, co je přímo na povrchu). Rtg. difrakční metoda kvantitativní fázové analýzy je tedy v tomto smyslu „někde na půl cesty“ mezi světelnou mikroskopií odraznou a (světelnou či elektronovou) mikroskopií prozařovací. A má tedy výhody i nevýhody obou těchto technik. Prohlížíme-li si výbrus horniny nebo histologický řez v polarizačním mikroskopu či kovovou fólii v transmisním elektronovém mikroskopu, dozvíme se jaké je (fázové) složení v celé tloušťce (prozařovaného) vzorku. Na rozdíl od toho, rtg. difrakce přináší informaci jen z několikamikronové tloušťky preparátu, a to ještě filtrovanou (rozuměj zkreslenou) tak, že komponenty směsné odezvy, které procházejí z větší hloubky jsou disproporcionálně potlačené vůči komponentám, které pocházejí z hloubky menší. Takové zkreslení (stejně jako ještě výraznější zkreslení, ke kterému dochází při pozorování světelným mikroskopem v uspořádání na odraz, kdy pozorujeme opravdu jenom to, co je na povrchu) není na závadu v případě, že struktura analyzovaného objektu je homogenní a isotropní. V opačném případě se pronikavost rtg. záření stává nevýhodou, protože omezuje hloubkovou rozlišitelnost při analýze hloubkového profilu fázového složení. K tomu účelu se dá využít rtg. záření různé vlnové délky (a tedy různé pronikavosti), různého úhlu dopadu a různé difrakční čáry (a tedy různé dráhy dopadajícího a difraktovaného paprsku ve vzorku), což se kombinuje s postupným odbrušováním (odleptáváním, odprašováním) povrchové vrstvy. Tím dostaneme soustavu integrálních rovnic a jejím řešením pak hledaný hloubkový profil fázového složení (jehož jest intensita záření určité vlnové délky, dopadajícího na vzorek pod určitým úhlem a difraktujícího pod nějakým zvoleným difrakčním úhlem při určité tloušťce odbroušené vrstvy, funkcionálem) [46].
Kvantitativní rtg. difrakční fázová analýza je velmi potřebná a užitečná technika. Jako taková se znamenitě uplatňuje v celé řádě přírodovědných i technických oborů. Není však příliš přesná (neurčitost jejích výsledků se pohybuje mezi
1-10 % absolutními). To je způsobeno tím, že intensity difrakčních linií, z nichž se při kvantitativní fázové analýze vychází, jsou vedle fázového složení ovlivněny také ještě reálnou strukturou. Reálná struktura je totiž velice proměnlivá a její vliv na intensity difrakcí lze jen obtížně kvantifikovat a odlišit od vlivu, který má na intensity difrakčních linií fázové složení.
Poděkování
Děkuji Ministerstvu školství, mládeže a tělovýchovy České republiky za finanční podporu v rámci projektu výzkumu a vývoje č. LNOOBO84, při jehož realizaci tato práce vznikla.
Literatura