Comparison of performance of universal Machine Learning Interatomic Potentials
Z. Holman1, M. Khanore1, A. Klic1, J. Hlinka1, J. Drahokoupil1, P. Marton1
1Institute of Physics of the Czech Academy of Sciences, Prague, Czech Republic
2Institute of Mechatronics and Computer Engineering, Technical University of Liberec, Czech Republic
Perovskites are materials with the crystal structure ABX₃ that can contain many different cations and anions. This flexibility allows their structural, electrical, and optical properties to be tuned and makes them important for a wide range of applications.
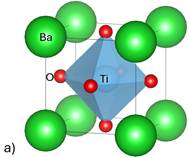
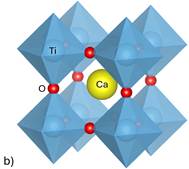
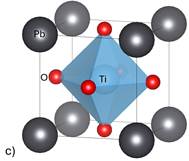
Figure 1. Structures of simulated perovskites a) BaTiO3 b) CaTiO3 c) PbTiO3
Understanding and improving their properties relies on computational modelling. Density Functional Theory (DFT) is commonly used as a reference method, but it is computationally expensive. Therefore, simplified approaches such as atomistic models, shell models, effective Hamiltonians, and phase-field methods are often used to study larger systems and processes like domain evolution and phase transitions. These models need parameters obtained either from experiments or from first-principles calculations. Recently, machine-learning interatomic potentials (MLIPs) have become a useful alternative because they can reach near-DFT accuracy while allowing relatively large and longe simulations of perovskite oxides.
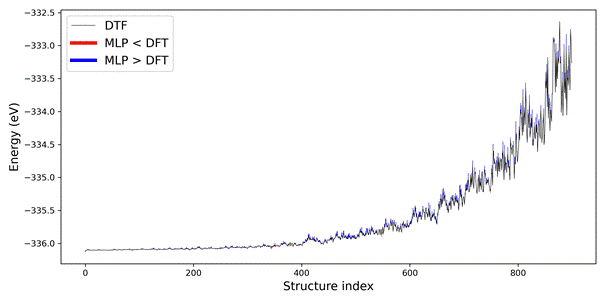
Figure 2. Energy difference between DFT and PET-MAD uMLIP per structure.
In this work we evaluate several variants of universal machine-learning interatomic potentials (uMLIP) [1,2,3,4]. We compare their accuracy of energy and forces predictions with DFT calculations. In this work we focused on performance of MACE, PET-MAD uMLIP.
Acknowledgements: The research was supported by Czech Science Foundation Grant No. 24-11275S.
[1] Ilyes Batatia, Dávid Péter Kovács, Gregor N. C. Simm, Christoph Ortner, and Gábor Csányi. Mace: Higher order equivariant message passing neural networks for fast and accurate force fields, 2023.
[2] Dávid Péter Kovács, Ilyes Batatia, Eszter Sára Arany, and Gábor Csányi. Evaluation of the mace force field architecture: From medicinal chemistry to materials science. The Journal of Chemical Physics, 159(4), 2023.
[3] Arslan Mazitov, Filippo Bigi, Matthias Kellner, Paolo Pegolo, Davide Tisi, Guillaume Fraux, Sergey Pozdnyakov, Philip Loche, and Michele Ceriotti. Pet-mad, a lightweight universal interatomic potential for advanced materials modeling, 2025.
[4] Cesare Malosso, Filippo Bigi, Paolo Pegolo, Joseph W. Abbott, Philip Loche, Mariana Rossi, Michele Ceriotti, and Arslan Mazitov. High-quality, high-information datasets for universal atomistic machine learning, 2026.