Exploring Local Atomic Environments using Universal Machine Learnig Potentials and Automated Structure Generation
J. Drahokoupil1,2,3, M. Lebeda1,2,3, Z. Holman1,2
1Fyzikální ústav, Akademie věd České Republiky, Na Slovance 2, 182 21 Praha 8, Česká Republika
2Katedra inženýrství pevných látek, Fakulta jaderná a fyzikálně inženýrská v Praze, České vysoké učení technické v Praze, Technická 4, 166 07 Praha 6 - Dejvice, Česká Republika
3Ústav fyziky, Fakulta strojní, České vysoké učení technické v Praze, Technická 4, 166 07 Praha 6 - Dejvice, Česká Republika
draho@fzu.cz
In the last few years, universal machine-learned interatomic potentials (UMLIPs) have entered the world of computer simulations, filling the gap between classical potentials and DFT calculations. Ideally, they achieve accuracy comparable to DFT but with a computational cost several orders of magnitude lower. Furthermore, their scaling is not cubic with respect to the number of atoms, making it possible to simulate structures containing hundreds of atoms even on a high-end desktop PC.
From the perspective of diffraction techniques, we usually observe an average structure where atomic positions may have partial occupancies. However, when describing structures for MD or DFT calculations, each atomic site must (with few exceptions) be either fully occupied or vacant. Therefore, if we want to simulate an alloy with a random atomic distribution, we must first generate such a configuration. Finding an arrangement that best mimics a truly random distribution is crucial for DFT calculations, where we typically work with small simulation cells. For this purpose, the concept of Special Quasi-random Structures (SQS) was introduced. While the ATAT mcsqs [1] program is commonly used to generate them, it isn't exactly user-friendly. To address this, our group developed SimplySQS [2], a tool designed to make this process much easier.
If the studied materials tend to exhibit atomic ordering, it is ideal to explore all possible configurations. This is practically feasible only for structures with a small number of atoms, as the number of combinations grows rapidly. A useful tool for generating various ordered structures is the Supercell [3] program, which also allows for the filtering of symmetry-equivalent structures.
To facilitate the use of universal potentials, our group is developing uMLIP-interactive [4]. This tool currently supports several universal ML potentials, including MACE, CHGNet, Nequix, SevenNet, Orb-v3, MatterSim, UPET, and GRACE. Through either a graphical interface or Python scripts, users can perform standard MD tasks such as energy calculation, geometry optimization, determination of elastic constants, phonon spectra calculations, and various dynamical simulations (NVE, NVT, NPT). Input files are supported in cif, POSCAR, lmp, or xyz (with lattice info) formats.
In this contribution, we will discuss three specific examples of use:
Preferential positions of gaseous elements in the "Gum Metal" alloy based on the Ti-23Nb-0.7Ta-2Zr composition.
When a small amount of oxygen (approx. 0.5%) is added to the Ti-23Nb-0.7Ta-2Zr alloy, it acquires remarkable properties: an elastic modulus close to 50 GPa combined with a strength exceeding 1000 MPa. Due to these unique characteristics, the alloy is referred to as "Gum Metal." Our research focused on identifying the preferential positions of interstitial atoms within this structure for low concentrations of gaseous elements: O, N, C, and H.
First, we transformed a simple bcc cell into a supercell containing 250 atoms and searched for an SQS structure corresponding to the composition in atomic percent (185 Ti, 58 Nb, 5 Zr, 2 Ta). From three independent runs, we obtained three distinct SQS structures. Within these structures, there are 750 octahedral and 1500 tetrahedral interstitial sites. We placed a single gas atom into each of these positions, performed geometry optimization, and statistically evaluated the entire data set. The results clearly show that O, N, and C prefer the larger octahedral sites, whereas H prefers tetrahedral sites. Furthermore, we found that for these interstitial atoms, a Ti-rich environment is energetically favorable, while an Nb-rich environment is unfavorable, see Fig. 2.
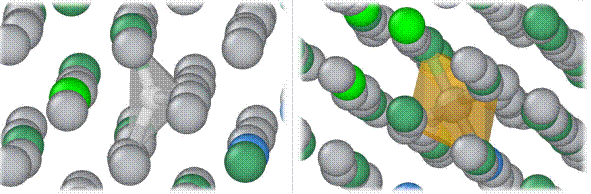
Figure 1. Stable tetrahedral position for a hydrogen atom (left) and stable octahedral position for a carbon atom (right) in the Ti-23Nb-0.7Ta-2Zr alloy.
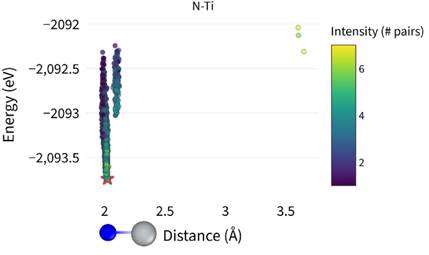
Figure 2. Energy dependence on the local environment of nitrogen atoms. It is evident that nitrogen atoms prefer environments dominated by titanium (Ti) atoms.
Hydrogen atom positions in Mg2Ni.
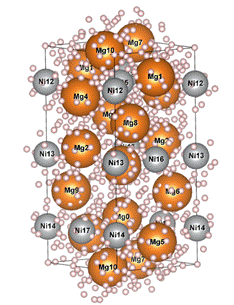
The compound Mg2Ni, sometimes enriched with rare earth elements, is a highly promising material for hydrogen storage, where the target stable phase is Mg2NiH4. Unfortunately, the saturation process can slow down in certain cases, making it difficult to load further hydrogen into the material. Understanding which sites hydrogen prefers within the original Mg2Ni structure is of great interest, and not only from a diffusion perspective. For low hydrogen concentrations, diffraction techniques do not always provide a clear answer regarding the occupied positions. Therefore, we attempted to determine the hydrogen sites by calculating the energy for various potential locations. Using the Point-Defect-Generator [5], another tool being developed in our team, we performed a grid search to identify all potential hydrogen positions (see Fig. 3a). Subsequently, we created a structure for each possibility containing a single hydrogen atom and performed geometry optimization using a universal potential. We then placed an additional hydrogen atom into the lowest-energy structure, and so on.
|
a) |
b) |
|
Figure 3. a) The structure Mg2Ni of with available interstitial sites for potential hydrogen localization (small pink spheres). b) Energies of individual structures containing two hydrogen atoms. |
|
Structure prediction of Mn – Hf – O.
The motivation for this research was the attempt to synthesize the MnHfO3 phase, which, according to theoretical calculations, should exhibit interesting magnetic properties. Preparation in powder form was unsuccessful, resulting instead in separated Mn2O3 and HfO2 phases. Consequently, we attempted to prepare samples in the form of thin films. In this case, we obtained a cubic Hf/MnO₂ phase. EDS analysis revealed that the oxygen concentration corresponds to an occupancy of 0.75. To verify whether the oxygen vacancies are a result of low oxygen concentration in the reservoir during thin-film growth, or if they are energetically driven by the specific Mn/Hf ratio, we again employed a universal potential to calculate the energies for various compositions and atomic distributions.
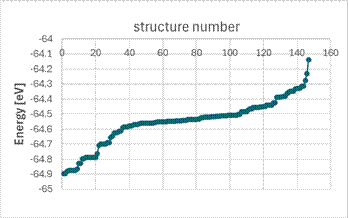
Starting from a cubic HfO2 structure, we replaced 50% of the Hf with Mn and considered various vacancy concentrations. Using the Supercell program, we generated all possible atomic configurations on appropriately sized supercells. With a finite (and in our case, small) number of atoms, the structure no longer maintains cubic or pseudo-cubic symmetry. To simulate real structures where cubic symmetry emerges statistically over a large number of atoms, it is appropriate to apply certain constraints. Fig. 4 shows the formation energy of the Hf/MnO₂ structure as a function of oxygen vacancy concentration under different constraints during geometry optimization. It is evident that, regardless of the constraints applied, an oxygen occupancy around 0.75 is energetically favorable. Furthermore, it can be seen that the local ordering also has a significant impact on the total energy.
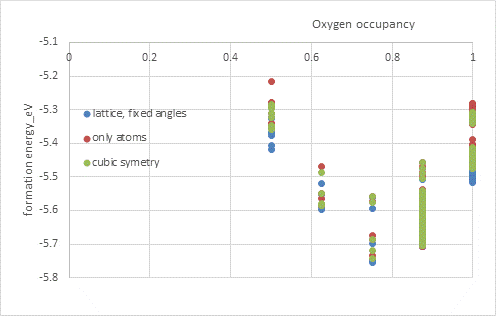
Figure 4. Formation energy of the Hf/MnO₂ structure as a function of oxygen vacancy concentration. Simulations were performed for various atomic arrangements within the cubic HfO2 structure. The Supercell program was used to generate the individual configurations.
[1] https://axelvandewalle.github.io/www-avdw//atat/
[2] https://atat-sqs.streamlit.app/
[3] https://orex.github.io/supercell/
[4] https://github.com/bracerino/uMLIP-Interactive
[5] https://xrdlicious-point-defects.streamlit.app/