The use of machine learning interatomic potentials for crystal structure solution verification
M. Hušák
1University of Chemistry and Technology, Prague, Technická 5, 166 28 Praha 6 – Dejvice
husakm@vscht.cz
Introduction
Correctly solved crystal structure should be in agreement with experimental data and its geometry should be located in a local minimum on a Potential Energy Surface (PES). The idea to verify crystal structure by comparison with DFT calculation results was introduced already 12 years ago [1,2]. Machine Learning Interatomic Potentials (MLIP) offer a significantly faster alternative to DFT as a tool for theoretical crystal structure calculation with comparable accuracy and correctness. The geometry optimized structures based on UMA(S) MLIP (parameterised on OMC25 dataset) [3] were used to verify 216 911 selected structures from Cambridge Structure Database (CSD). The targeted was to detect issues with both original X-ray experiments interpretation as well as with the MLIP functionality.
Methodology
Before the structure validation we had pre-filtered from CSD 6.0 a subset of structures, for which it had sense to run the test (See Table1). We had rejected structures with disorder, used MLIP non supported elements, voids, incorrect space group and to old structures measured on historical devices. Current MLIP do not support well electrostatic long-range forces so we had rejected structures with charged atoms as well. From used GPU memory limits reasons structures with unit cell volume more than 4000Å3 were not investigated. See Table 1 for the pre-filtering results.
Table 1. Result of structures pre-filtering
|
Rejection reason |
Structures rejected based on given test |
Structures left after test |
|
Total structures in CSD 6.0 dataset |
|
1371757 |
|
Presence of other elements than supported one ( H,C,N,O,S,P,F,Cl,Br,I) |
853820 |
517937 |
|
Disordered structure |
90765 |
427172 |
|
Future not satisfied: No error, 3D coordinates present, single crystal data |
32241 |
394931 |
|
Not published after 2004 |
95698 |
299233 |
|
Ionized molecules or charged atoms present |
41768 |
257465 |
|
Volume over 4000Å3 |
17725 |
239740 |
|
3D coordinates not present (not covered by previous check) |
4 |
239736 |
|
Solvent accessible volume over 40Å3 |
16463 |
223273 |
|
Incorrect space group or super cell detected |
1128 |
222145 |
|
Incorrect element valence detected |
3981 |
218164 |
|
Detected atoms marked by "?" indicating disorder |
1253 |
216911 |
The final MLIP calculation was running 4 months in parallel on 2 computers with NVIDIA RTX PRO 4000 Blackwell graphic card equipped by 24GB/ GDDR7 memory. Execution was done almost exclusively in GPU on 8 960 CUDA cores. The processing and descriptors calculation was driven by our proprietary software checkCIF-DFT.
The original verification method [1,2] was primary based on the experimental and DFT results comparison by Cartesian displacement (RMSCD) descriptor only. RMSCD is de-facto a RMSD modified to be able to compare atomic positions in different unit cells. It was already mentioned [1] this descriptor was not able to clearly separate e.g. totally artificial fraud structures from correctly solved one. We had tested several other descriptors to separate problematic structures from correct ones. Really problematic structures can be detected efficiently by bond-breaking (see Table 2 and Figure 1).
Table 2. Issues detected during the CSD 216911 structures test
|
Problem detection reason |
Structures rejected based on given test |
Structures left after test |
|
Initial dataset |
|
216911 |
|
Non existing (Unknown) element present |
1 |
216910 |
|
CIF space group does not correspond to original CIF |
1 |
216909 |
|
Bond breaking (not including H-atom) |
466 |
216443 |
|
Bond breaking (H-atom involved) |
1400 |
215043 |
|
RMSCD over 1.0 Å |
167 |
214876 |
|
RMSCD over 0.25 Å |
3167 |
211709 |
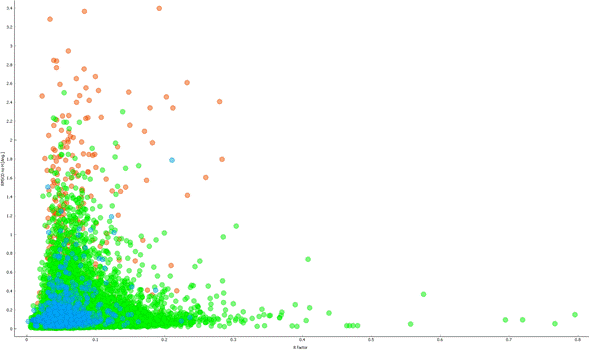
Figure 1. R factor versus RMSCD (hydrogen atoms excluded). green - no bond breaking, blue - only bonds including hydrogen break, red - breaking of bonds not including hydrogen
Results
We had manually inspected 466 structures with bond breaking (hydrogen not involved) and 167 structures with RMSCD higher than 1.0 Å. The inspection was based on comparison with original X-ray data, higher level full DFT calculation and inspection of the original publications. We had identified several types of errors with the CSD deposited data: element type assignment miss-match, missing hydrogen, incorrect hydrogen placement or incorrect experimental information in the CIF (pressure typically). We had identified issues with the used MLIP PES generation as well - incorrect proton transfer and pure results for elements not often present in teaching dataset. Most of the PES related issues were related to the PBE+D3 functional used for OMC25 dataset generation. PBE-D3 shows known errors related to ion transfer. A better teaching set utilizing e.g. r2SCAN-MBD meta-GGA functional will be more suitable for future work. In rare situation we had detected as problematic correctly solved structures with non-usual futures. .
Conclusions
During examination of our results we had identified issues with multiple structures deposited in CSD. It should be noted that based on our finding the use of CSD as reliable source of molecular geometry information is questionable. Marking the problematic structures in the database with some other flack than "no error" will be suitable. On the other side we had identified issues with the used MLIP as well - incorrect proton transfer and incorrect geometry for bond types not occurring often in the OMC25 dataset. The computational time required for single structures sets is typically less then one minute - the described methodology is suitable for routine structure checks.
1. J. Streek, M. A. Neumann, Acta .Cryst., B66, (2010), 544.
2. J. Streek, M. A. Neumann, Acta .Cryst., B70, (2014), 1020.
3. Wood, B. M., Dzamba, M., Fu, X., Gao, M., Shuaibi, M., Barroso-Luque, L., Abdelmaqsoud, K., Gharakhanyan, V., Kitchin, J. R., Levine, D. S., Michel, K., Sriram, A., Cohen, T., Das, A., Rizvi, A., Sahoo, S. J., Ulissi, Z. W. & Zitnick, C. L. (2026). Uma: A family of universal models for atoms. https://doi.org/10.48550/arXiv.2506.23971