Optické vlastnosti koloidních soustav
(fyzikální princip metody
měření velikosti částic a zeta potenciálu)
Optické
vlastnosti koloidních soustav jsou silně závislé zejména na
fyzikálních vlastnostech koloidních částic - zejména jejich velikosti,
elektrické vodivosti a vlastní absorpci světla látkou tvořící disperzní fázi. Z
jevů spojených s interakcí světla s koloidními soustavami poskytují nejvíce
informací rozptyl a absorpce světla, proto se dále
budeme zabývat pouze těmito jevy. Čistý
rozptyl světla nastává za podmínky, že vlnová
délka světla λ je
podstatně větší,
než koloidní částice v soustavě (o
poloměru r), tedy platí-li podmínka λ >>
r. Je-li tomu naopak, dochází přednostně
k odrazu světla a pak pozorujeme zákal (hrubě
disperzní soustavy). Klasickou
teorii statického rozptylu světla
za uvedené podmínky λ >>
r vypracoval Rayleigh. Podle
této teorie na základě představy částice jako oscilujícího dipólu, vyzařujícícho pohlcenou světelnou energii do všech stran beze změny vlnové délky (rozptyl světla), závisí rozptýlená intenzita světla Iθ
jak na elektrických vlastnostech rozptylující částice (polarizovatelnost α), tak na vlnové délce světla a rovněž na pozorovací vzdálenosti R a pozorovacím úhlu θ:
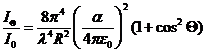
V praxi se ovšem metoda měření statického rozptylu světla pro komplikovaný výpočet rozměru rozptylujících částic neujala, ale je využívána pro určení molekulové hmotnosti makromoleku-lárních látek, protože pro tuto veličinu získáváme ze statického rozptylu světla poměrně jednoduchou rovnici:
![]()
kde c je koncentrace částic v soustavě a K představuje konstantu rozptylu (ta je určena vlastnostmi měřícího zařízení a vlnové délky světla). Uvedený vztah platí pouze pro ideálně se chovající soustavy, pro reálné se situace komplikuje a rozptýlená intenzita závisí na koncentraci částic komplikovaněji (vyjádřeno pro rozptylový úhel 90º):
![]()
kde veličina B’ představuje druhý viriální koeficient soustavy (korekce na její neineideální chování). Celé měření se v reálu provádí pro různé rozptylové úhly a pro různé koncentrace, přičemž výsledná hodnota molekulové hmotnosti se určuje z extrapolace na nulové hodnoty obou proměnných (Zimmova metoda). Pro částice nesplňující podmínku λ >> r, ale srovnatelným velikostí s vlnovou délkou použitého světla byla vytvořena nadstavbová teorie (Debye), která ukazuje, že rozptylová křivka již není zrcadlově symetrická a že původní Rayleighovy rovnice stačí upravit vynásobením vhodným korekčním faktorem. Pro ještě větší částice vytvořil novou teorii Mie, který ukázal, že závislost rozptýlené intenzity na úhlu pozorování je již značně nelineární a není prakticky použitelná pro výpočet rozměru rozptylující částice. Navíc závislost na vlnové délce použitého světla rovněž neodpovídá Rayleigho teorii a příslušná mocnina je menší než 4. Další komplikace přináší elektrická vodivost částic disperzního prostředí, kdy dochází vlivem existence povrchového plasmonu (kolektivní oscilace vodivostních elektronů ve fázi s elmg. vlněním - světlem) k masívní absorpci světla při určitých vlnových délkách, což opět narušuje výše uvedené vztahy mezi rozptýlenou intenzitou světla a dalšími parametry soustavy.
Při obecném rozptylu světla je světlo, rozptýlené od různých částic, v náhodné fázi, takže nedochází k jeho interferenci. Při použití laseru je světlo koherentní a po jeho interakci s částicemi k interferenci dochází. Protože se malé částice v kapalině pohybují díky Brownovu pohybu, mění se vzdálenost, kterou musí urazit rozptýlené světlo k detektoru. Rozptýlené vlnění může interferovat v závislosti na vzdálenosti mezi částicí a detektorem. Výsledkem jsou fluktuace intenzity rozptýleného záření okolo průměrné hodnoty intenzity. Z fluktuací intenzity se získá korelační funkce, která představuje vztah mezi průměrem intenzity v čase (t + τ) a v čase t. Při velkém τ (zpoždění) jsou I(t) a I(t+τ) na sobě nezávislé, zatímco při velmi krátkém zpoždění na sobě závislé jsou. V případě kulovitých částic stejných velikostí lze korelační funkci vyjádřit v jednoduchém exponenciálním tvaru:
![]()
kde parametr τc je úměrný difúznímu koeficientu D částic:
![]()
kde Q je vlnový vektor.
Vynesením ln g(τ) proti τ se získá τc. Hydrodynamický poloměr částic R je, za použití Stokes-Einsteinovy rovnice pro difúzní koeficient (D = kT/6πηa), dán vztahem:
![]()
V praxi se měří τc při různých úhlech rozptylu (různých hodnotách Q) a vynáší se 1/τc proti Q2, čímž se získává D a odtud R.
Dynamický rozptyl světla
Metoda dynamického rozptylu světla (DLS), nazývaná rovněž jako foton korelační spektroskopie (PCS) je v současné době široce používána pro stanovení velikosti koloidních částic. Obvykle jsou výsledky v přiměřeně dobré shodě s výsledky elektronového mikroskopu. Určité nesrovnalosti ve výsledcích mohou být přisouzeny buď rozdílu mezi hydrodynamickým poloměrem (který může zahrnovat solvatační obal či naadsorbovanou vrstvu na povrchu částic) a skutečným poloměrem nebo změnám ve velikosti částice vznikajícím při přípravě vzorku pro elektronový mikroskop (vysoušení) či změnám pocházejícím od ozařování elektrony. V případě polydisperzních systémů se musí vzít do úvahy pohyb částic rozdílných velikostí a korelační funkce má poté složitější formu.
Získaná průměrná velikost koloidních částic je vážena podle páté mocniny (tzv. Z-average), takže velké částice jsou v této hodnotě velmi nadhodnoceny a dokonce se může u některých přístrojů stát, že frakce malých částic se úplně ztratí ve velké intenzitě záření rozptýleného na větších částicích. Rozsah metody je oproti sedimentačním metodám výrazně jiný - dolní mez se dnes pohybuje okolo 0,5 nm a maximální pak okolo 3 μm (větší částice již nepodléhají Brownovu pohybu).
Přítomnost částic disperzní fáze a široce rozvinutého fázového rozhraní v disperzních soustavách podmiňuje zvláštní ráz jejich elektrických vlastností a především vznik tzv. elektrokinetických jevů. Podobně jako malý jednoduchý ion kolem sebe vytváří iontovou atmosféru protiiontů, tak se i kolem nabité koloidní částice seskupují malé ionty opačného znaménka, takže na povrchu této částice vznikají dvě nabité vrstvy, tzv. elektrická dvojvrstva.
První představy o struktuře elektrické dvojvrstvy
přinesl H. Helmholtz. Představoval si ji jako dvě k sobě přiléhající rovnoběžné desky nabitého kondenzátoru. Jedna deska je tvořena ionty fixovanými na povrchu pevné
fáze. Ionty určují
potenciál této vrstvy. Druhá deska je tvořena opačně nabitými ionty, které pochází z kapalného
disperzního prostředí.
Problémem této teorie je fakt, že Helmhotzův model dvojvrstvy předpokládal její
nehybnost. To-není ale možné vzhledem k tepelnému
pohybu molekul disperzního prostředí.
Helmhotzova teorie byla překonána teorií difúzní
dvojvrstvy, kterou nezávisle na sobě vypracovali Goüy (1910) a Chapman (1913). Vážným nedostatkem
jejich teorie je fakt, že-se na ionty vytvářející dvojvrstvu pohlíží pouze jako na bodové náboje. To pak vedlo k výpočtu
nesmyslně velkých
koncentrací nábojů v
blízkosti fázového rozhraní. Korekci-provedl v roce
1924 Stern, který zavedl pojem tloušťka
adsorbované vrstvy δ (popř.-d), která odpovídá přibližně hodnotě iontových poloměrů.
V elektrické dvojvrstvě tak lze vymezit dvě základní části. Kompaktní část bližší k povrchu, kde
působí adsorpční síly, a vzdálenější difúzní část, kde lze tyto adsorpční síly zanedbat. Vzhledem k povrchovému náboji koloidních částic existuje potenciálový rozdíl mezi
jejich povrchem a roztokem. Lze rozlišit dva druhy
potenciálových rozdílů.
Prvním z nich je elektrochemický potenciál, jehož hodnota je dána celkovým
potenciálovým rozdílem mezi povrchem částice a objemem kapaliny. Je odpovědný za jevy spojené s
vedením elektrického proudu a za membránové potenciály. Druhým
potenciálem je elektrokinetický potenciál (ζ potenciál,
zeta potenciál), jímž se rozumí potenciálový rozdíl mezi objemem kapaliny a
tenkou vrstvou protiiontů poutanou k povrchu částice, tedy na rozhranní mezi kompaktní
a difúzní částí elektrické
dvojvrstvy. Podle Sterna je první vrstva, a s ní i několik vrstev protiiontů, přitahována k povrchu jak elektrostatickými, tak adsorpčními silami. Část protiiontů tedy zůstává v blízkosti povrchu,
ve vzdálenosti řádově 1 až 2 molekulových průměrů, což lze popsat modelem
deskového kondenzátoru. V této adsorpční vrstvě dochází k prudkému poklesu elektrického potenciálu. Zbylé
protiionty, nutné ke kompenzaci náboje iontů
určujících
potenciál, vytváří v
důsledku tepelného
pohybu difúzní část
elektrické dvojvrstvy. Ze schématu Sternova difúzního modelu elektrické
dvojvrstvy je zřejmé, že celkový
potenciálový rozdíl se skládá z poklesu potenciálu ϕδ difúzní
části dvojvrstvy a z rozdílu potenciálů (ϕ0 − ϕδ) mezi vrstvami, jež se
pokládají za desky modelového kondenzátoru. Po přídavku elektrolytu do systému se bude difúzní vrstva stlačovat a stále více protiiontů se ocitne v adsorpční vrstvě. Zeta potenciál se bude snižovat, až dosáhne
téměř nulové hodnoty.
Při
ředění systému se difúzní
vrstva naopak rozšiřuje
a zeta potenciál vzrůstá.
Elektrický náboj dvojvrstvy charakterizuje a
zároveň ovlivňuje stabilitu koloidních systémů. Zeta potenciál (ζ),
který odpovídá náboji difúzní části
dvojvrstvy, je právě mírou
tohoto náboje, přičemž název
elektrokinetický potenciál získal díky existenci tzv. elektrokinetických
jevů, tedy kinetických dějů způsobených vlivem elektrického pole na koloidní soustavu. Existují čtyři základní typy
elektrokinetických jevů -
elektroforéza, elektroosmóza,
sedimentační
potenciál a potenciál proudění. Všechny tyto jevy jsou současně využitelné pro
určení hodnoty elektrokinetického potenciálu, nejčastěji se používá
elektroforetických měření.
Elektroforéza je jevem, při kterém dochází k pohybu částic
disperzních fáze v elektrickém poli. Elektroforetická
rychlost v s jakou se částice disperzní fáze v elektrickém poli pohybují se u
většiny anorganických solů pohybuje mezi 2 a 4.10-4 cm2.V-1.s-1.
Samotný pohyb dispergovaných částic je pozorovatelný i přímo
v ultramikroskopu. Pro výpočet elektroforetické rychlosti odvodili P.
Debye a E. Hückel rovnici platící pro ideální kulovité
částice s poloměrem r, které se pohybují v elektrickém poli intenzity E/L (kde E je napětí vložené na kyvetu o délce L):
![]()
kde D představuje dielektrickou konstantu a η viskozitu disperzního prostředí. Tato rovnice tedy umožňuje zjištění zeta potenciálu koloidních částic z měření jejich elektrické pohyblivosti.
Schema
elektrické dvojvrstvy a jejího ovlivnění.
Tloušťka dvojvrstvy může být ovlivněna řadou faktorů. Nejdůležitější je vliv přítomnosti elektrolytů. Ionty z difuzní části elektrické dvojvrstvy, která se pohybuje s okolní kapalinou, přecházejí přes pohybové rozhraní do části lpící na tuhém povrchu. Stlačování dvojvrstvy má za následek pokles elektrokinetického potenciálu. Účinnost iontů při stlačování dvojvrstvy je dána jednak mocenstvím iontů, jednak jejich adsorptivitou; adsorpční vrstvu tvoří protiionty. Při dostatečné koncentraci vhodného elektrolytu je celá vnější vrstva stlačena za pohybové rozhraní; tuhý povrch nemá náboj, je dosaženo izoelektrického bodu. Jak tuhý povrch, tak kapalina jsou v tomto stavu elektroneutrální, elektrokinetický potenciál je roven nule a elektrokinetické jevy vymizí.
Elektrolyt obsahující ion schopný účasti v krystalové mřížce, může změnit hodnotu povrchového potenciálu (V0). Zbývající ion, jehož náboj je stejný jako náboj protiiontu, může stlačovat elektrickou dvojvrstvu. Při malých koncentracích elektrolytu převládá povrchový účinek iontu, který je schopen zabudovat se do krystalové mřížky. Při větších koncentracích převládá stlačování dvojvrstvy. Proto se přídavkem stále větších množství elektrolytu ζ-potenciál nejprve zvyšuje, dosáhne maxima a potom klesá. Přídavek elektrolytu může vyvolat i změnu znaménka koloidních částic.
Indiferentní elektrolyty, které neobsahují ionty podobné iontům krystalové mřížky koloidní částice, snižují ζ-potenciál v důsledku zvýšení iontové síly roztoku. Vzrůstá koncentrace protiiontů ve vnější vrstvě a dvojvrstva se stlačuje. Většími přídavky elektrolytu je možno dosáhnout stlačení dvojvrstvy až za pohybové rozhraní a částice se při pohybu vůči kapalné fázi jeví jako elektricky neutrální (ζ = 0, izoelektrický bod).
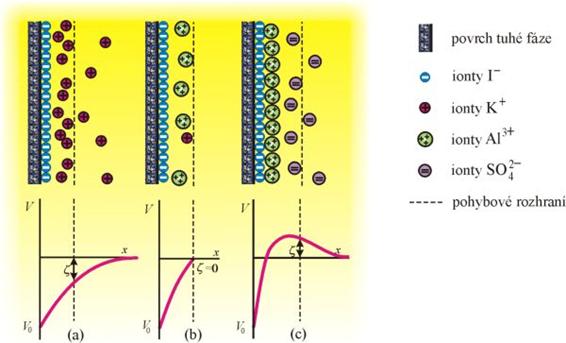
Vícemocné ionty mohou i změnit znaménko ζ-potenciálu. Např. při přídavku síranu hlinitého k solu AgI stabilizovanému jodidem draselným (ionty I– tvoří první vrstvu na povrchu AgI, vnější vrstva obsahuje ionty K+ - obr. 1a) dochází k výměně málo vázaných K+ iontů za ionty hlinité. Při určité koncentraci síranu hlinitého je ζ = 0 a systém je v izoelektrickém bodě - obr. 1b. Při dalších přídavcích hlinité soli vzniká na první, záporně nabité vrstvě adsorpční vrstva iontů Al3+ (obr. 1c), která má kladný náboj a přitahuje k sobě záporné ionty - vzniká nová dvojvrstva. ζ-potenciál změní znaménko, křivka ζ(c) prochází extrémem. Vnější, záporně nabitá vrstva se dalšími přídavky opět stlačuje za pohybové rozhraní a ζ-potenciál se blíží nule.
Literatura
1.
P. C. Hiemenz : Principles of
Colloid and Surface Chemistry, Marcel Dekker 1986
2.
M. Takeo : Disperse Systems,
Wiley-VCH, Weinheim 1999
3.
Z. Samec, Elektrochemie, skripta
PřF UK, 1999