The artificial protein Octarellin challenges crystallographers and modellers
M. Figueroa1,2, M. Sleutel3, D. Maes3, C. Van de Weerdt1
1GIGA-Research, Molecular Biology and Genetic Engineering Unit, University of Liège, Liège, Belgium
2Biochemistry and Molecular Biology Department, University of Concepcion, Chile
3Structural Biology Brussels, Vrije Universiteit Brussel, Belgium
dommaes@vub.ac.be
The aim of de novo design proteins, often called the inverse protein folding problem, is to find amino acid sequences compatible with a given protein tertiary structure. Solving the inverse folding problem questions our understanding of sequence-structure relationships in proteins. Despite impressive successes in de novo protein design, designing a well-folded protein of more than 100 amino acids remains a challenge.
We will discuss the artificial protein Octarellin designed to adapt the TIM-barrel fold1,2. Crystallization was only successful after the creation of stable complexes with antibodies from camelids (nanobodies) and alfa-repeat (αRep) proteins3,4. As it turns out, the experimental X-ray structure deviates considerably from the idealized design, failing even at fold level. The experimental (αβα) sandwich architecture bears some resemblance to a Rossman-like fold instead of the intended TIM-barrel fold. This surprising result gave us a unique and attractive opportunity to test the state of the art in protein structure prediction. We tested 13 automated webservers for protein structure prediction and found none of them to predict the actual structure. More than 50% of them predicted a TIM-barrel fold.
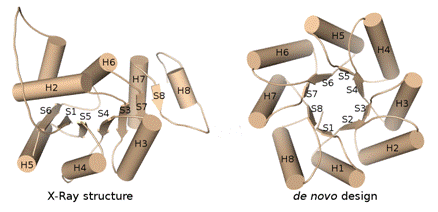
Figure 1. Comparison of the de novo designed and the X-ray structure from Octarellin.
Note that the expected strand 2 (S2), helix 1 (H1), and the majority of the helix 8 (H8) are missing in the X-ray structure.
1. F. Offredi et al, J Mol Biol, 325, (2003), 163
2. M. Figueroa et al., Plos One, 8 , (2013), e71858.
3. M. Figueroa et al., accepted for publication in Journal of Structural Biology.
4. E. Pardon et al., Nat Protoc., 9, (2014), 674.