Mechanism of inhibition of hGLUT1 is conserved between cytochalasin B and phenylalanine amides
K. Kapoor1, J. Finer-Moore1, B. Pedersen1, 2, L. Caboni1, R. Hillig3, P. Bringmann4, I. Heisler3, T. Müller3, H. Siebeneicher3 and R. Stroud1
1Department of Biochemistry and Biophysics, University of California, San Francisco, CA, 94158
2Department of Molecular Biology and Genetics & Aarhus Institute of Advanced Studies, Aarhus University, Aarhus DK-8000 Denmark
3Bayer HealthCare Pharmaceutical, Berlin/Wuppertal, Germany.
4 Bayer HealthCare LLC, 455 Mission Bay Blvd., San Francisco, CA 94158
kapoor@msg.ucsf.edu
Human glucose transport (hGLUT1) belongs to a family of homologous sugar
transporters or co-transporters found in both prokaryotes and eukaryotes [1].
It is a uniporter that transports glucose from the extracellular matrix into
cells [2]. Cancerous cells have an acutely increased demand for energy leading
to increased levels of hGLUT1 [3]. This highlights hGLUT1 as an important
prognostic indicator in several cancer types [4, 5], as well as a potential new
target for therapeutic inhibitors. Here we present inhibitor-bound inward-open
structures of wild-type hGLUT1 crystallized with three different inhibitors: cytochalasin
B, a nine-membered bicyclic ring fused to a 14-membered macrocycle, which has
been described extensively in the literature of hGLUTs [6] and two novel
phenylalanine amide derived inhibitors. Despite very different chemical
backbones, all three compounds bind in the central cavity of the inward-open
state of hGLUT1 and all binding sites overlap the glucose-binding site (Figure
1). The inhibitory action of the compounds was determined for hGLUT family
members, hGLUT1-4, using cell-based assays, and compared to homology models for
these hGLUT members. This uncovered a probable basis for the observed
differences in inhibition between family members.
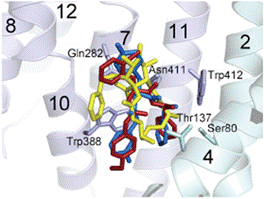
Figure 1. Overlapping inhibitor-binding sites in hGLUT1.
We pinpoint regions of the hGLUT proteins that can be targeted to achieve isoform selectivity, and show that these same regions are used for inhibitors with very distinct structural backbones. These structures provide an important structural insight for the design of more selective inhibitors for hGLUTs and hGLUT1 in particular. Thus our results emphasize that modulation of glucose import by hGLUTs should focus on making good interaction points for compounds and that the actual chemical backbone of the inhibitor is of less importance.