Use of X-ray crystallographic data for computational modelling of receptor-ligand interactions: design of steroidal inhibitors of breast and prostate cancer cell growth
E. Petri1, A. Ćelić1, J. Plavša1, M. Marinović1, S. Bekić1, O. Klisurić, M. Sakač2
1Department of Biology and Ecology, Faculty of Sciences, University of Novi Sad, Serbia
2SDepartment of Chemistry, Biochemistry and Environmental Protection
edward.petri@dbe.uns.ac.rs
Hormone-sensitive tumours, such as breast or prostate cancers, are leading causes of death, and manipulation of steroid signaling is an effective treatment. X-ray structures of steroid receptors have been solved in complex with anti-cancer drugs (e.g. tamoxifen); and steroid modifying enzymes have been solved in complex with steroidal anti-tumour drugs (e.g. exemestane, Abiraterone). Although X-ray crystallography is essential for structure-based drug design, the vast chemical space prevents experimental analysis of all interacting compounds. To design novel anti-tumour steroidal compounds, we use X-ray crystallographic data from protein-ligand complexes as templates for molecular dynamics, Monte Carlo and molecular docking simulations using freely available resources. X-ray structures of proteins in complex with steroidal drugs used to treat hormone-dependent cancers were chosen: estrogen receptor, androgen receptor , aromatase (CYP19), 17α-hydroxylase (CYP17), 17b-HSD family enzymes and aldo-keto reductases (AKR1Cs) [1]. We use in silico methods to ask: Is it possible to reliably model new receptor-ligand complexes using X-ray structural data from known receptor-ligand structures. Simulation results are correlated with in vitro and anti-proliferation tests using human cancer cells. In general, in silico computational methods appear to predict the molecular targets of steroidal compounds, and can refine protein X-ray crystallographic data to model new ligand binding geometries (Fig 1) [2-4]. However, modelling ligand binding starting from apo structures is likely to fail. Moreover, for flexible ligand sites, X-ray structures of chemically similar ligand complexes appear necessary, combined with refinement by molecular dynamics or Monte Carlo. Thus, additional X-ray structures of proteins bound to diverse steroidal ligands are necessary for design of improved anti-cancer steroidal compounds.
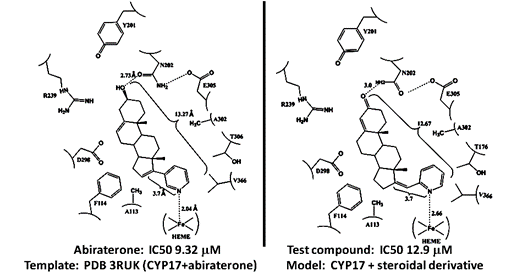
Figure 1. Example: modelling binding by new steroidal derivatives using X-ray structural data (CYP17 + anti-cancer drug Abiraterone). Predicted binding energies correlate with anti-proliferative activity.
1. N. DeVore, E. Scott, E. E. Nature, 482, (2012), 116-119.
2. J. Ajduković, E. Djurendić, E. Petri, O. Klisurić, A. Ćelić, M. Sakač, K. Gaši, Bioorg. & med. Chem., 21, (2013), 7257-7266.
3. K. Penov-Gaši, A. Oklješa, E. Petri, A. Ćelić, E. Djurendić, O. Klisurić,...M. Sakač, MedChemComm, 4, (2013) 317-323.
4. A. Nikolić, E. Petri, O. Klisurić, A. Ćelić, D. Jakimov, E. Djurendić,...M. Sakač, Bioorg & med chem, 23, (2015), 703-711.
We thank the Ministry of Education and Science of the Republic of Serbia for financial support (Grant No. 172021).