INSIGHTS INTO THE CALCIUM-INDUCED CONFORMATIONAL TRANSITION OF DOMAIN III IN ANNEXIN V USING CPR MODELISATION
J. Sopkova1,3,
S. Fischer2, C.
Guilbert1, A.
LewitBentley3
& J. C. Smith1
1 Section de
Biophysique des Protéines et des Membranes, Departement de
Biologie Cellulaire et Moleculaire, CEASaclay, 91191
Gif-sur-Yvette Cedex, France
2 Computational and Structural Chemistry, Hoffman-La
Roche/Pharma Research, CH-4070 Basel, Switzerland
3 LURE, Centre Universitaire Paris-Sud, Bâtiment 209D,
91405 Orsay Cedex, France
Annexin V belongs to a family of proteins that share large structural homologies and biochemical characteristics. In particular, all these proteins have been found to interact with negatively charged phospholipid membranes in a calcium-dependent manner at neutral pH [1] and to cell membranes [2]. To date, the exact biological function(s) of these proteins remain(s) unknown but several observations have suggested that they can be involved in physiological processes involving interaction with constituents of cellular membranes like in signal transduction, in exocytosis, as regulators of phospholipase A2 activity or as membrane effectors and also in the cytoskeleton as regulatory elements.
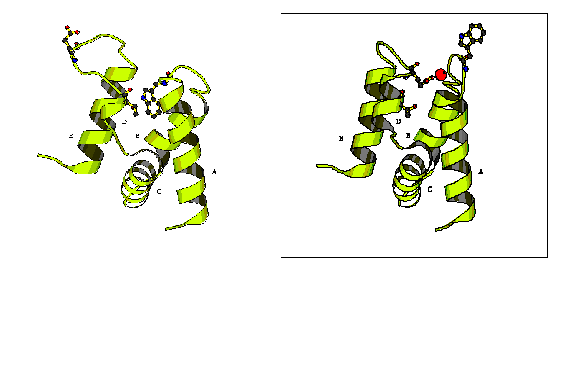
Annexin V, due to its potential anticoagulant activity, has been the most studied member of the annexin family and its 3D structure was the first to be solved [3]. To date several different Xray structures of annexin V and its mutants, prepared under different crystallisation conditions, have been obtained. It was shown that when the calcium ion is not bound in domain III (we call this the 'product' structure), the loop connecting helices A and B lies in the centre of the antiparallel helix bundle [3] (see Figure). When the calcium ion is bound in domain III, other crystal forms are observed (the 'reactant' structure) [4] (see Figure). The AB loop moves in a direction extending helices A and B, thus bringing Trp 187 onto the surface of the protein. The D-E loop moves to a position opposite to the form without the calcium site in domain III, thus allowing glutamic acid 228 to approach the calcium site and to complete the calcium ligands. The RMSD over all nonhydrogen atoms of domain III between reactant and product structures is 4.1A. This conformational difference is significant, both in terms of its amplitude and in terms of the number of residues involved (80 amino acids, i.e. one entire domain). For illustration, the Ca atom of Glu228 moves about 12 A, and the Ca atom of Trp187 moves about 9A.
While crystallography can provide valuable information on different equilibrium conformations of a protein, the experimental characterisation of pathways between conformations is much more difficult. Various forms of spectroscopy can sometimes be used to obtain transition rates, but the pathway itself, due primarily to its non-equilibrium rarely-populated nature, cannot be directly determined. A further difficulty encountered is in conformational transitions involving many degrees of freedom, such as in the present case, for which there is no guarantee that a single pathway will be taken. The above arguments indicate that to obtain information on conformational pathways in proteins it is necessary to make use of simulation methods. Global methods that refine the reaction path as a whole, like the conjugate peak refinement method (CPR) [5-6] can be used. The CPR method finds the minimum energy path which follows the valleys and crosses the saddlepoints of the high dimensional energy surface. It finds this path without requiring an a priori choice of which degrees of freedom constitute the reaction co-ordinate.
One of the problems during this simulation was the introduction of solvatation effect corrections; we tested several models. The pathway refined using a weak solvatation correction (the dielectric constant was dependent on the distance) corresponds to a potential energy barrier of about 30kcal/mol. Along the pathway we observed trans-cis-trans transitions of two peptide bonds situated in the proximity of Trp 187: Gly 188 and Asp 190. Detailed analysis of the transition state showed that the charged residues on the protein surface made hydrogen bonds between them, and thus constrained domain III. Therefore, we repeated the simulation introducing stronger corrections of solvatation effects. For this purpose, we rescaled the partial atomic charge to account for solvent shielding effects, which were determined from a Poisson-Boltzman calculation of the electrostatic potential, in which the solvent is treated as a high-dielectric continuum. In the new refined pathway we observed a decrease of the potential energy barrier by about 25 kcal/mol to an energy barrier of about 5 kcal/mol. In this pathway we again observed the trans-cis-trans transition of a peptide bond, however only for one residue - Asp 190. The necessity to have constant atomic composition along the CPR pathway makes the inclusion of explicit solvent molecules in the calculation impossible. The explicit introduction of water molecules is easily possible in the case when the conformational change occurs in the protein interior, and when the protein volume changes minimally. However, in the present case the conformational change concerns the movement of loops on the protein surface, and the volume of domain III therefore changes significantly between the reactant to the product, from 8861 A3 to 7856 A3 (i.e. about 13%).
Independently of the corrections on the solvatation effects
used during the pathway refinement we can divide visually the
path into the four principal phases. The first is a rearrangement
of helix D. The second is a stretching of the DE loop, which
adopts an intermediate position between the reactant and product
structures. In this way a space is created for the insertion of
Trp187. The third step is the gradual insertion of Trp187 into
the protein cavity between the helices of domain III. The
conformational change is finished by the complete turning of the
DE loop to the position corresponding to the product structure.