STRUCTURES OF 5-R-3-ACETYLIMINO-3H-[1,2]-DITHIOLE-4-CARBONITRILE AND THEIR 3-THIOACETYLIMINO ANALOGUES
Jaromír Marek, Michal Cajan, Richard Cmelik & Pavel Pazdera
Faculty of
Science, Masaryk University, Kotlá_ská 2, CZ 611 37 Brno, Czech
Republic;
e-mail : cajan@chemi.muni.cz;
marek@chemi.muni.cz
Keywords : trithiaazapentalene skeleton, crystal
structure, ab-initio quantum chemistry modelling, FTIR
spectra
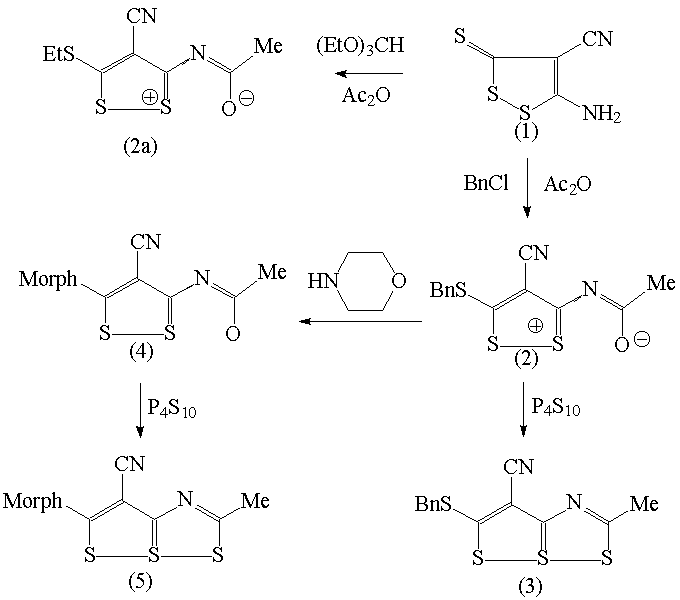
When carbonitrile (1) described in [1] reacts with triethyl orthoformate in the presence of Ac2O as the accelerator of the reaction, the result is not expected ethoxymethyleneimino derivative, but the compound (2a) C8H8N2OS3 containing methyl, ethyl and cyano groups. The surprise for us was that FTIR and 13C NMR spectra of (2a) do not contain characteristics typical for C=O group. That is why we realized also reaction of (1) with in sequence Ac2O and BnCl with resulting product (2) too and why we prepared single crystals of (2) suitable for X-ray analysis by vapour-diffusion method from chloroform as dissolvent and petroleum ether as reservoir. In the following reaction the compound (2) reacted with P4S10 and resulting compound (3) was isolated in crystalic form from chloroform. Morpholinolysis of (2) gives (4) and exchange of acetyl oxygen into (4) by P4S10 proceed to formation of (5).
Crystals of (2) and (3) were studied by X-ray structure analysis and the results were confronted with quantum chemistry ab-initio calculations of compounds (2)-(5) performed at HF and B3LYP level of theory. The X-ray structure of (2) reveals that experimental C-O distance 1.249(4) is slightly greater than C-O distance typical for carboxyle group (the average distance in the Cambridge Structural database [2] is 1.225 A) and then the result of ab initio calculation (1.2025 A). From the bond length and from relative big difference of the charges on O and neighbour S atoms (-0.48 vs. 0.39) we deduce that the sake of missing carboxyle vibration in the experimental spectra is in the electrostatic interaction between S and O atoms. The agreement between maxima on observed FTIR and theoretical spectra in range 500-1600 cm-1 was excellent.
The crystal data for (2)
were C13H10N2OS3,
Mr=306.41, Monoclinic crystal system, space group P21/c,
a=10.344(2), b=8.356(2), c=16.318(2) A, b=107.24(2)°, V=1347.1(4) A3 , Z=4, rx=1.511
Mg.m-3 , m=0.541 mm-1, T=150.0(1) K, l=0.71073 A=MoKa,
reflections collected / unique 11844 / 2357 [R(int) = 0.0507], R
indices [I>2s(I)] R1=0.0374, wR2
= 0.0848 and the largest diff. peak and hole 0.376 and -0.444 e.A-3,
the data for (3) are C13H10N2S4,
Mr=322.47, Triclinic crystal system, space group P-1, a=7.750(3),
b=9.623(5), c=10.035(4) A, a=74.49(4)°, b=78.78(3)°, g=73.72(4) °,
V= 686.4(5) A3 ,
Z=2, rx=1.560 Mg.m-3 , m=0.677 mm-1,
T=150.0(1) K, l=0.71073 A=MoKá, reflections collected /
unique 2520 / 2367 [R(int) = 0.0331], R indices [I>2s(I)] R1=0.0543,
wR2 = 0.1823 and the largest diff. peak and hole 0.869
and 0.574e A-3