 |
 |
ON THE CONFORMATION OF TWO 1,4-DIARYL-2,3-DINITRO-1,3-BUTADIENES
Carlo Dell'Erba, Angelo Mugnoli, Marino Novi, Giovanni Petrillo and Cinzia Tavani
Dipartimento di Chimica e Chimica Industriale, Universita di Genova,
and Centro C.N.R.- C.S.C.C.C.A., Via Dodecaneso 31, I-16146 Genova, Italy.
E-mail: mugnoli@chimica.unige.it
Keywords: Conformation, Ab initio MO calculations,
Weak interactions, C-H...O bonds
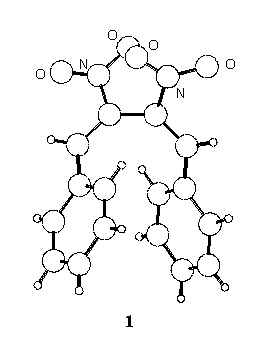
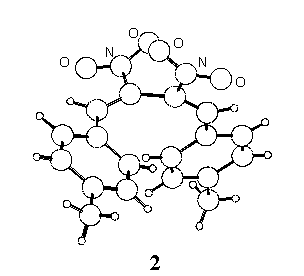
Following our interest in the field of conjugated nitro-unsaturated systems [1,2] the crystal structure of two derivatives, 1,4-diphenyl- (1 ) and 1,4-di-p-tolyl-2,3-dinitro-1,3-butadiene (2) has been determined.
Crystal data:
1, monoclinic, P21/c, Z=4, a=10.152(2), b=8.937(2), c=16.204(5) A, b=93.15(2)o, V=1467.9(6) A3, current R=0.05 over about 2100 reflections;
2, monoclinic, P21/c, Z=4, a=12.095(3), b=7.337(2),
c=19.195(4) A, b = 107.12(2)o, V=1627.9(7) A3,
current R=0.04 over about 2000 reflections. All H atoms located by difference syntheses
and refined isotropically.
In the crystal state the two conformations are quite different, the C=C-C=C torsion angle being synclinal (55o) in 1 and anticlinal (121o) in 2, where the molecule assumes a more extended shape.
Starting from the experimental results, a geometry optimization has been accomplished for the isolated molecules with ab initio MO calculations (HF/6-31G*, 354 basis functions for 1, 392 for 2). Both models converged (very slowly for 2) to essentially the same geometry, with a C=C-C=C torsion angle of 70o.
The intramolecular distance between the ring centroids is 4.27 A in 1 and 6.82 A in 2, and 5.01 and 5.05 A respectively in the isolated molecules; in 1 the planes of the phenyl rings define a dihedral angle of 37o, which reduces to 32o for the calculated conformation.
In the crystals the shortest intermolecular distances between ring centroids of 1 and 2 (4.89 and 4.86 A, respectively) correspond to centrosymmetric pairs; the (parallel) mean planes of the involved rings are stacked at 3.58 and 3.69 A for 1 and 2 respectively, but in both cases the parallel displacement is too high [3] to allow p - p stacking interactions to exert appreciable effects.
As ascertained in previous known cases [4,5], we believe that the presence of two
intermolecular C-H···O hydrogen bonds in 2 (C-H 0.98 and 0.95 A, C···O 3.40
and 3.37 A, H···O 2.44 and 2.46 A, C-H···O 166.2 and 159.5o,
respectively) is largely responsible for the conformation of 2 in the crystal
state.
|
|